
中国水稻主产区白叶枯病菌致病型分析及近等基因系鉴别寄主的构建
2022-12-01 07:36冯爱卿汪聪颖张梅英陈炳封金奇陈凯玲汪文娟杨健源苏菁曾列先陈深朱小源
中国农业科学 2022年21期
冯爱卿,汪聪颖,张梅英,陈炳,封金奇,陈凯玲,汪文娟,杨健源,苏菁,曾列先,陈深,朱小源
中国水稻主产区白叶枯病菌致病型分析及近等基因系鉴别寄主的构建
冯爱卿,汪聪颖,张梅英,陈炳,封金奇,陈凯玲,汪文娟,杨健源,苏菁,曾列先,陈深,朱小源
广东省农业科学院植物保护研究所/广东省植物保护新技术重点实验室,广州 510640
【目的】分析中国不同稻区白叶枯病菌(pv.)致病型,建立近等基因系鉴别寄主,为白叶枯病菌群体结构田间实时准确监测、抗性品种应用以及抗病育种提供科学依据。【方法】利用中国鉴别寄主、IR24以及15个抗白叶枯病近等基因系等共21个鉴别寄主,采用人工剪叶接种方法,对2018—2021年采自广东、广西、海南、浙江、湖南、辽宁、云南共7个省(自治区)的954个单菌落分离菌株进行致病型测定,探明白叶枯病菌致病型种类、分布及毒性分化;基于测试菌株与15个近等基因系及IR24的抗感互作,应用主成分因子分析法,开展近等基因系与病菌互作的变量因子分析,构建白叶枯病菌致病型近等基因系鉴别寄主;基于抗病基因与测试菌株的抗感反应,分析抗病基因聚合效应。【结果】954个测试菌株在中国鉴别寄主上鉴定出11个致病型,包括SRRRR(I)、SSRRR(Ⅱ)、SSSRR(Ⅲ)、SSSSR(Ⅳ)、SSRRS(V)、SRSRR(Ⅵ)、SSSSS(Ⅸ)、SSSRS(新型1)、SRSRS(新型2)、SRSSS(新型3)以及SSRSS(新型4),占测试菌株的比率分别为11.53%、4.82%、7.34%、6.18%、7.23%、1.05%、59.96%、1.57%、0.10%、0.10%、0.10%。Ⅸ型菌作为致病性最广的强毒菌系已上升为华南和长江中下游湖南和浙江稻区的优势致病型,西南稻区的云南以Ⅳ型菌为主,东北稻区的辽宁以I型菌为主。15个水稻抗白叶枯病近等基因系对954个菌株的抗感性分析结果表明,测试的15个近等基因系可分为5种类型,第Ⅰ类为高感基因系,包括IRBB1、IRBB2、IRBB10、IRBB11、IRBB4;第Ⅱ类为中感基因系,包括IRBB3、IRBB203、IRBB14;第Ⅲ类为中抗基因系,包括IRBB8、IRBB13;第Ⅳ类为抗病基因系,有IRBB21;第Ⅴ类为高抗基因系,包括IRBB5、IRBB7、CBB23、GDBB23;测试菌株中,出现可侵染抗病基因的有42个、的有34个、的有31个。对以白叶枯病近等基因系为主的16个品种(系)与954个菌株组成的抗感互作变量数据矩阵进行因子分析,以解释总变量>85.0%为界,提取出8个主成分因子,组建了以近等基因系为主的10个品种(系)组成白叶枯病菌近等基因系鉴别寄主,按其对变量方差贡献大小,这些寄主分别为IRBB10()、IRBB4()、GDBB23()、IRBB5()、IRBB7()、IRBB21()、IR24()、IRBB13()、IRBB3()、金刚30;新鉴别寄主可将954个测试菌株划分为55个致病型,对测试稻区的白叶枯病菌菌株表现出较好的鉴别力。基因聚合联合抗性分析表明,不同抗病基因聚合对病菌的抗性频率有一定的提升,不同抗病基因对测试菌株的抗性具有一定的互补性。【结论】监测稻区的白叶枯病菌系趋向多样化,毒性分化明显,强毒菌系Ⅸ型菌在部分稻区已上升为优势致病型,侵染及等广谱抗性基因的菌株有上升趋势;抗病基因聚合可拓宽品种对病原菌系的抗性谱,是培育广谱抗性品种的有效途径;近等基因系鉴别寄主的建立与应用可为白叶枯病发生流行的精准监测以及田间实时预警提供技术支撑。
水稻白叶枯病菌;致病型;近等基因系;鉴别寄主;抗性
0 引言
【研究意义】白叶枯病是影响我国乃至世界水稻生产最重要的细菌性病害之一。该病最早于1884年在日本发现,20世纪30年代在我国江苏、浙江就有发生,目前发生足迹已遍布我国各水稻主产区,尤其以南方沿海稻区发生较为严重[1-2]。白叶枯病具有病菌来源广、增殖速度快、传播途径多、危害程度严重等特点,一旦显症,常规方法已较难防治。国内外各种实践已证明利用寄主抗性是控制该病害最经济、有效的方法,可实现该病害的可持续治理。针对白叶枯病菌(pv.,)毒性不断变异的特点,监测、研究病菌致病型发生消长动态,建立精准的鉴别体系,对准确、实时监测田间白叶枯病菌群体结构和病害预警,以及对白叶枯病抗病育种和抗性品种应用均具有重要的科学意义和应用价值。【前人研究进展】水稻与白叶枯病菌的互作符合基因对基因关系,被认为是研究植物-病原物互作的典型模式系统[3-4],因此,监测鉴定白叶枯病菌致病型、致病机制以及挖掘与之相应的抗病基因已成为国内外研究的重点。在白叶枯病菌致病性研究方面,1958年,日本久原重松等最早提出在白叶枯病菌中存在着致病性不同的小种[5]。1985年,NODA 等利用金南风群、黄玉群、RantilEmas群、早爱3群和Java群5个鉴别寄主群检测出日本白叶枯病菌的新致病型Ⅶ[6];至20世纪80年代末,在日本白叶枯病鉴别系统中,利用Kinmaze、Kogyoku、Rantai-Emas、Wase Aikoku、Java、Elwee、Heen Dikwee、IR8共8个鉴别寄主鉴定了ⅠA、ⅠB、Ⅱ、ⅢA、ⅢB、Ⅳ、Ⅴ、Ⅵ、Ⅶ共9个致病型[7]。1987年国际水稻研究所通过菲律宾菌系在IR8、IR20、IR1545、DV85、Cas209 5个鉴别品种上的反应型和菌系分化情况,将白叶枯病菌致病型正式称之为生理小种,初步鉴定出6个生理小种[8]。伍尚忠等[9]运用国际水稻研究所及日本的鉴别品种对不同国家的菌系进行了鉴定,发现这两套鉴别品种对不同菌系群的反应各不相同,不同国家和地区之间的病菌致病类型也不同,并提出各个国家应选择适宜本地区的鉴别体系来分析本国病原菌的发生消长状况。1985—1988年中国病理学家通过全国的统一试验,按照当时的菌株选出了一套适合中国水稻白叶枯病菌致病性分析的5个鉴别品种金刚30、Tetep、南粳15、Java14、IR26,并确认了SRRRR(Ⅰ型)、SSRRR(Ⅱ型)、SSSRR(Ⅲ型)、SSSSR(Ⅳ型)、SSRRS(Ⅴ型)、SRSRR(Ⅵ型)、SRSSR(Ⅶ型)共7个致病型[10]。利用这套中国鉴别品种2001年王春连等[11]、2005年曾列先等[12]分别在云南省、广东省水稻白叶枯病菌中各检测出1个新类型RRRSR(Ⅷ型)、SSSSS(Ⅸ型)。白叶枯病菌致病型SSRRS(Ⅴ型)、SSSSS(Ⅸ型)的发现表明该病菌毒性正在向更广致病谱方向演变,已经攻克了当时认为抗性最强的含有的品种,这是缘于80—90年代大面积种植含有品种,导致病原菌在持久、过度的选择压力和环境共同作用下发生的变异。陈深等[13]通过对华南稻区水稻白叶枯病菌的致病性变异动态分析发现,强致病菌系V型已替代Ⅳ型发展为华南优势致病菌系。近等基因系是研究病原菌与抗性基因互作及致病性演变的理想材料,1982—1987年,Ogawa等[14]采用统一菌系通过系统鉴定,将日本与国际水稻所鉴定的16个抗性基因修正为14个,统一编号为—。1991年在此基础上培育了一套以IR24为轮回亲本的籼稻近等基因系IBBBI—RBBI4、IRBB2I[15]。我国章琦等育成了一套以沈农1033为轮回亲本的粳稻近等基因系CBB2、CBB3、CBB4、CBB7、CBB12、CBB14[16]和以金刚30为轮回亲本的粳稻近等基因系CBB23[17]。目前,被鉴定的白叶枯病抗病基因至少已有46个[18-19]。同时随着水稻抗白叶枯病近等基因系的育成,国内外学者已开始利用这些近等基因系鉴别系统开展白叶枯病菌致病小种变异的研究[4,13,20-23],重建新的小种鉴别体系已成为我国白叶枯病研究者的共识。【本研究切入点】随着病原毒性变化,适时调整建立鉴别力强的新鉴别体系,对精准监测白叶枯病菌致病型发生消长动态、白叶枯病抗病育种以及利用抗性品种控制病害意义重大。如何利用白叶枯病近等基因系科学客观地建立新的鉴别寄主,目前的研究及其方法尚有待完善。【拟解决的关键问题】利用中国鉴别寄主以及国内外构建的抗白叶枯病近等基因系,通过对2018—2021年采自我国不同稻区共954个单孢分离菌株进行致病性测定分析,了解我国水稻产区白叶枯病菌致病型分布状况及其毒性分化;并利用系列抗性基因与来源不同的病原广泛互作的抗感变量信息,应用因子分析法寻找互作变量的关键因子,构建鉴别力好、冗余性低的白叶枯病菌近等基因系鉴别寄主,探明白叶枯病菌的毒性分化和优势致病型,抗病基因聚合对菌株抗性的聚合效应,为实现更精准的抗病品种合理布局及进一步选育新型抗白叶枯病新品种(组合)提供科学依据。
1 材料与方法
试验于2018—2021年在广东省农业科学院植物保护研究所试验基地发病环境适宜的试验田中完成。
1.1 病原菌材料、分离及保存
测试菌株:于2018—2021年采集,当年采集当年测定,来自我国不同生态区的广东、广西、海南、湖南、浙江、辽宁、云南共7个省(自治区)的954个单菌落分离菌株(表1)。其中广东省450个菌株,来自广州、丰顺、封开、佛冈、高州、广宁、化州、怀集、江门、蕉岭、揭西、雷州、连山、龙川、四会、台山、新会、新兴、信宜、兴宁、阳春、阳江、阳西、珠海24个市(县);广西133个菌株,来自容县、博白、平果、都安北、藤县、崇左、横县、靖西8个市(县);海南省14个菌株,来自海口、保亭等;湖南省14个菌株,来自浏阳等;辽宁省26个菌株,来自丹东、清源等;云南省11个菌株,来自保山等;浙江省306个菌株,来自忂州、常山、江浦、椒江、金华、龙游、宁波、瑞安、台州、温岭、仙居、诸暨12个市(县)。2018年测试193个菌株,2019年测试288个菌株,2020年测试179个菌株,2021年测试294个菌株。
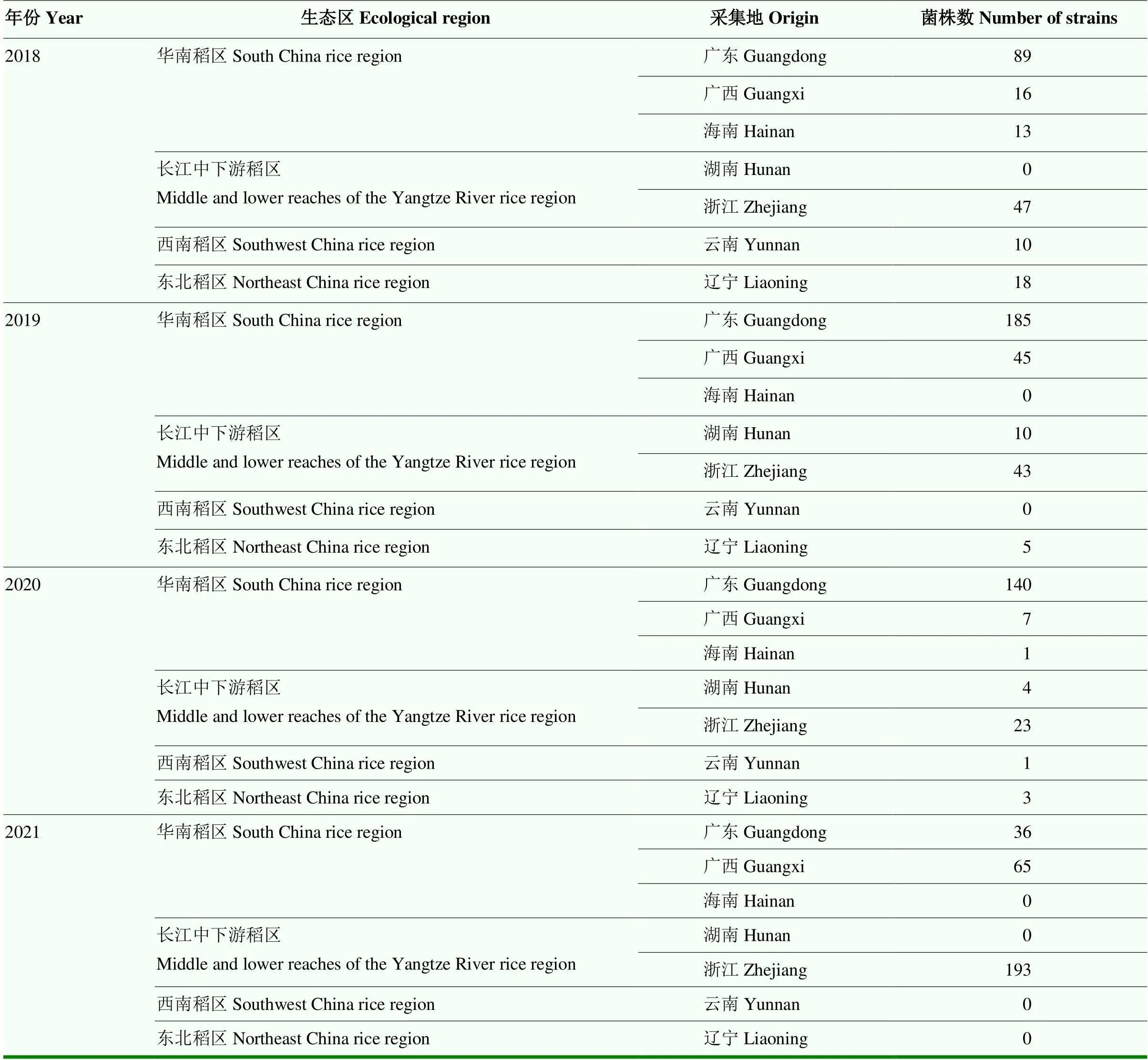
表1 测试菌株来源
菌株分离及保存:主要采用组织分离法和稀释分离法先将病原菌从病组织分离出来,再提纯和保存。组织分离法:先在病健交界处选取新鲜病组织,然后用无菌水冲洗,选用75%酒精表面消毒,放入0.1%升汞消毒1—1.5 min,再用无菌水冲洗3次,稍晾干后用剪刀修剪,去除多余的健康组织,最后将消毒过的组织块放在胁本哲氏马铃薯半合成(Wakimoto potato dextrose agar,WPDA)固体斜面培养基上28—30℃培养。稀释分离法:按上述方法取样消毒后,将新鲜病组织研碎加入适量无菌水浸泡10—15 min,稀释成不同梯度的菌悬液,适量倒入35℃左右的液体培养基摇匀凝固后,放置于28—30℃培养箱培养,也可将菌悬液划线或涂布于固体WPDA培养基上培养。病菌提纯:用划线法提纯单菌落菌株,移入试管斜面培养基放入4℃或应用甘油放入-70℃保存备用。
1.2 鉴别寄主、种植与管理
鉴别寄主21个。其中中国白叶枯病鉴别寄主5个:CBBD1(金刚30)、CBBD2(特特普)、CBBD3(南粳15)、CBBD4(爪哇14)、CBBD5(IR26,含)。抗白叶枯病近等基因系15个:IRBB1()、IRBB2()、IRBB3()、IRBB203()、IRBB4()、IRBB5()、IRBB7()、IRBB8()、IRBB10()、IRBB11()、IRBB13()、IRBB14()、IRBB2I()、CBB23()、GDBB23(),其中CBB23由中国农业科学院作物科学研究所章琦研究员提供;GDBB23是以CBB23为供体、IR24为受体及轮回亲本,经过6次回交及4次自交发展而成,含的抗白叶枯病近等基因系,由广东省农业科学院植物保护研究所选育;其余近等基因系从国际水稻研究所(IRRI)引进。感病对照:IR24。
种植与管理:鉴别品种编号后,按正常季节播种育秧,秧龄期早稻30 d,晚稻25 d以内。按编号顺序,每个品种插一列,单本移植,两次重复,株行距18 cm×24 cm,接种菌株间间隔40 cm以上。鉴别品种种植于广东省农业科学院植物保护研究所试验基地发病环境适宜的试验田中,土壤肥力中等、均衡,试验期间不施用其他杀菌剂,其他栽培管理措施一致。
1.3 参试菌株的致病型(小种)测定
在水稻孕穗期,人工剪叶接种法(用剪刀蘸取菌液,剪去叶尖2—3 cm,蘸一次剪一片叶)接菌。接种菌株在WPDA平板上复壮,在培养基上28—30℃培养48 h后,用无菌水稀释菌种,接种菌液浓度为3×108cfu/mL。每一菌株在每个鉴别品种上接种稻株20片叶(以剑叶为主),设两次重复。
接种后21 d调查,用病斑面积(长度)占叶片面积(长度)百分率作为抗感反应参数,参照全国白叶枯病菌菌系研究病级标准调查和记录。病斑长度达到接种叶长的1/4或病斑面积大于接种叶面积的20%为感病,病斑长度小于接种叶长的1/4或病斑面积少于接种叶面积的20%为抗病[10],根据各菌株在各鉴别寄主上的抗感反应,划分小种(近等基因系)和菌系致病型(中国鉴别寄主)。
1.4 华南水稻白叶枯病菌致病型变异分化动态分析
将2018—2021年华南稻区的菌株在中国鉴别寄主上的致病型检测结果,结合本研究室历史研究数据,比较分析1999—2021年华南稻区病菌致病型变异分化动态。本研究室1999—2014年共测定了276个菌株[13]、2015—2017年分别测定了68、50、60个菌株,基于各自鉴定的致病型及其发生频率,与本研究所得结果比较,分析华南稻区白叶枯病菌优势致病型的变异动态。
1.5 数据处理与分析
采用Microsoft Excel 2010进行调查原始数据处理。采用IBM SPSS Statistics Version 20软件[24],对近等基因系与病菌互作变量进行因子分析和聚类分析,互作数据分为1(感病反应)和0(抗性反应)。
2 结果
2.1 接种结果的有效性
接种21 d调查,蘸菌剪叶接种中国鉴别寄主金刚30高感品种发病充分,均产生白叶枯病典型症状(图1),各菌株在高感品种金刚30上病斑长度均达到接种叶长的1/4 或病斑面积大于接种叶面积的20%以上。说明各接种菌株均有一定的毒性且发病环境条件适宜,均达到试验要求,本结果有效。

图1 不同年份、不同白叶枯病菌菌株接种金刚30症状
2.2 测试菌株在中国鉴别寄主上的致病反应
2.2.1 测试菌株在中国鉴别寄主上的致病型 根据954个测试菌株在中国鉴别寄主CBBD1(金刚30)、CBBD2(特特普)、CBBD3(南粳15)、CBBD4(爪哇14)、CBBD5(IR26)上的抗感反应归类,鉴定出SRRRR(I型)、SSRRR(II 型)、SSSRR(III 型)、SSSSR(IV 型)、SSRRS(V型)、SRSRR(Ⅵ型)、SSSSS(Ⅸ型)、SSSRS(新型1)、SRSRS(新型2)、SRSSS(新型3)、SSRSS(新型4)11个致病型。其中Ⅸ型菌株有572个,发生频率最高,为59.96%,表明Ⅸ型菌作为致病性最广的强毒菌系已上升为这些地区的优势致病型,且白叶枯菌系趋向多样化(表2)。另外,监测地区的水稻白叶枯病菌2018—2021年每年均以Ⅸ型菌发生频率最高(表3)。
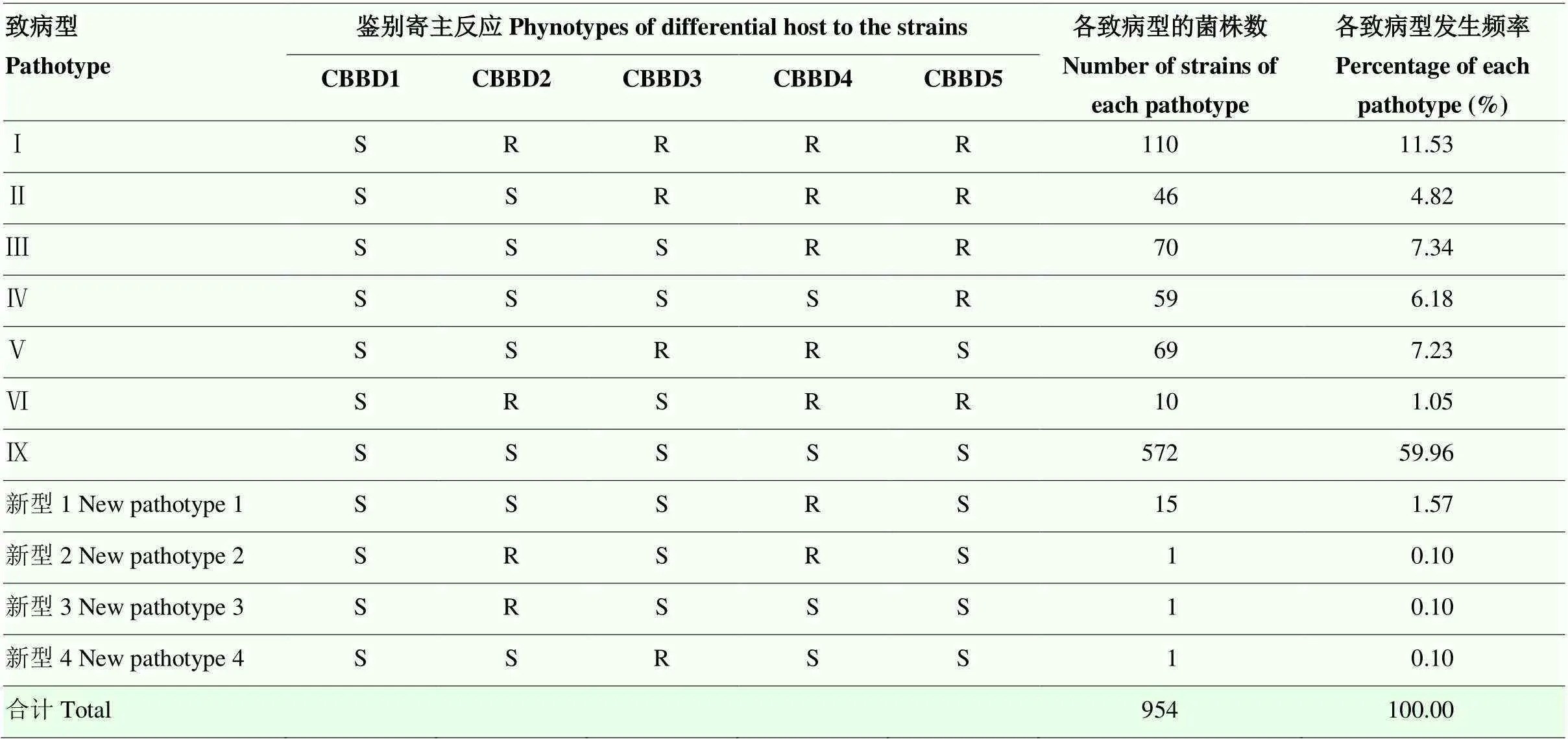
表2 测试菌株在中国鉴别寄主上的致病反应
R:抗病Resistant;S:感病Susceptible
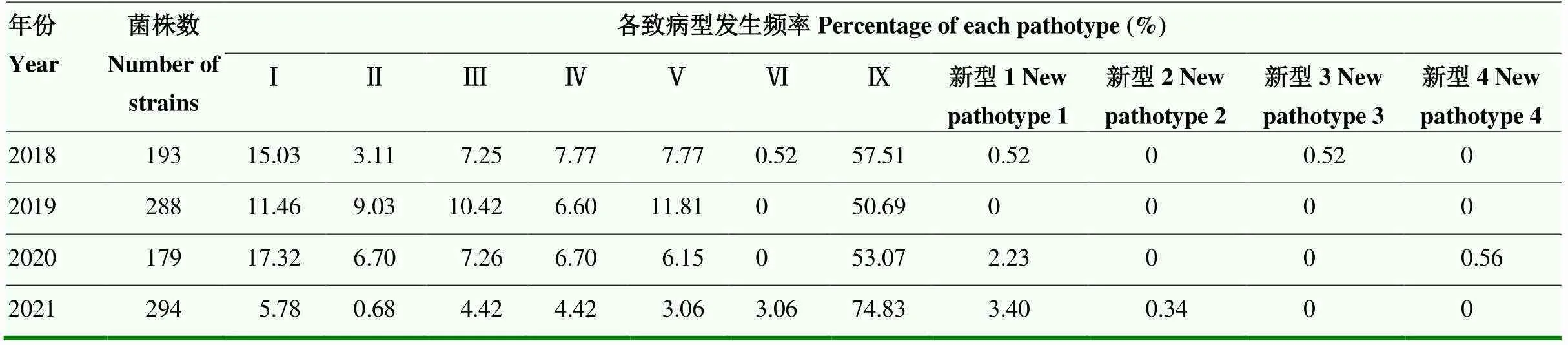
表3 2018—2021年中国7省(自治区)水稻白叶枯病菌致病型和发生频率
2.2.2 不同生态稻区的致病型分布 954个测试菌株分别来自4个生态稻区。其中华南稻区的597个白叶枯病菌株在中国鉴别寄主上可分为10个致病型,最为丰富,Ⅸ型菌为该稻区的优势致病型,发生频率为55.61%;长江中下游稻区测试了320个菌株,可分为9个致病型,较为丰富,Ⅸ型菌亦为该稻区的优势致病型,发生频率为74.38%;西南稻区测试的11个菌株中,可分为4个致病型,Ⅳ型菌发生频率最高,为72.73%;东北稻区测试了26个菌株,可分为5个致病型,I型菌发生频率最高,为65.38%(图2)。综上所述,本研究测试菌株中,华南稻区、长江中下游稻区均以Ⅸ型菌为主,西南稻区以Ⅳ型菌为主,东北稻区以I型菌为主。
2.2.3 华南水稻白叶枯病菌1999—2021年致病型变异分化动态 历史研究数据与本研究数据比较发现,1999—2017年华南稻区主要有I、Ⅱ、Ⅲ、Ⅳ、V和Ⅸ这6个致病型,2018年后陆续有Ⅵ型和其他新的致病型出现,虽然频率不高,但病菌分化更为多样(表4)。2004年以前华南稻区主要以Ⅳ型菌为优势致病型;而2014—2017年,V型菌基本成为了华南稻区的优势致病型,发生频率在20.59%—27.40%;2004年发现的强毒菌系Ⅸ型菌发生频率持续上升,由2004年的5.70%升至2021年的72.28%,已成为华南稻区的优势致病型(表4)。
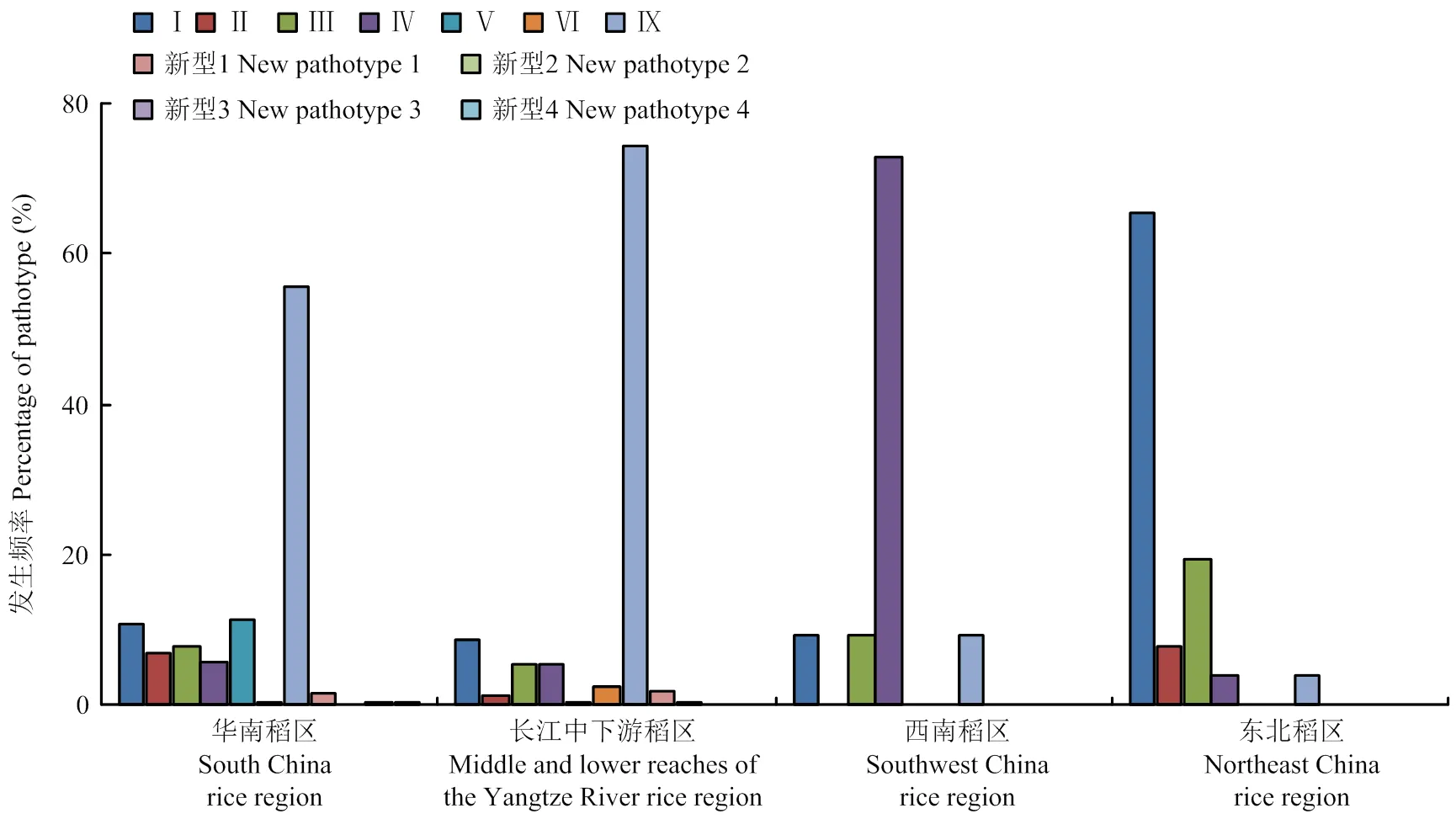
图2 我国不同水稻生态区白叶枯病菌致病型分布
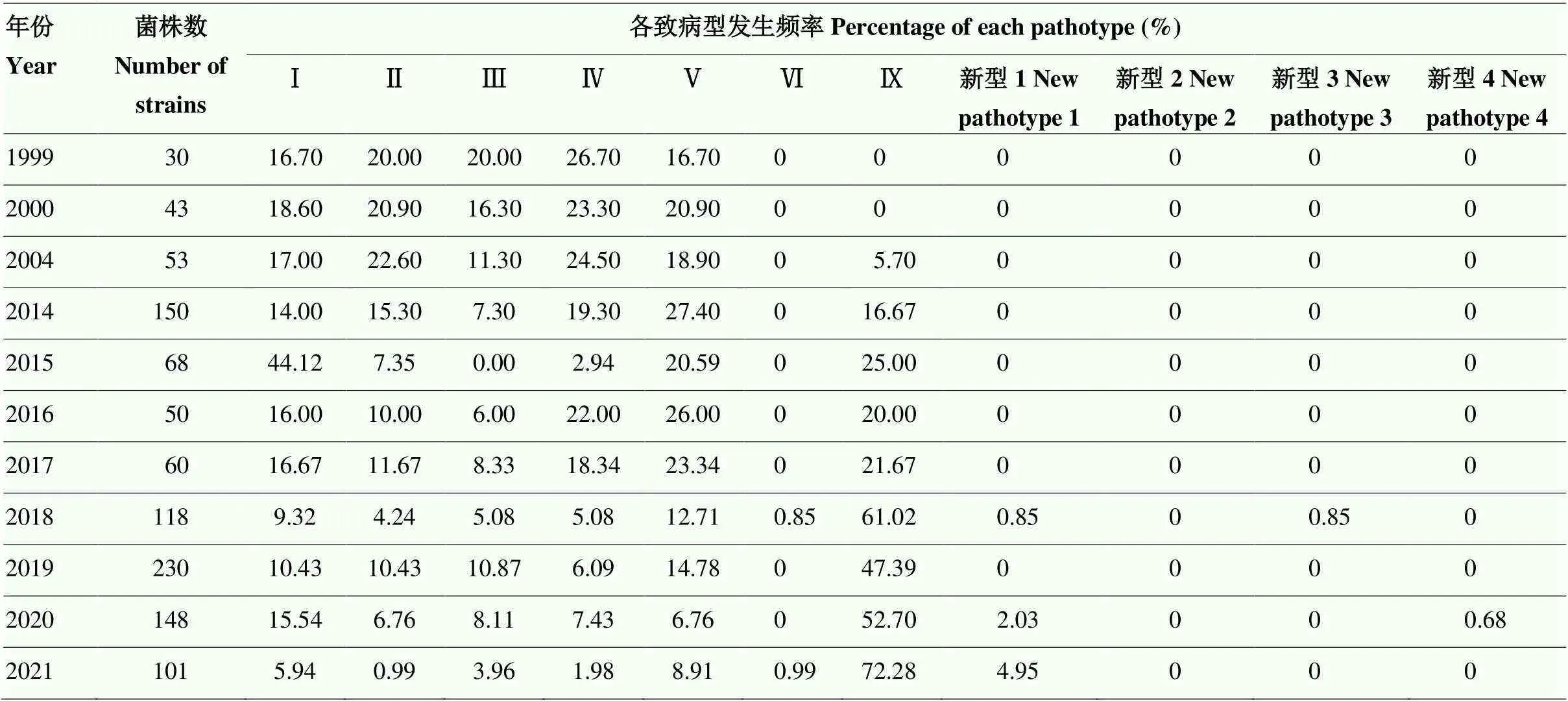
表4 1999—2021年华南水稻白叶枯病菌致病型及发生频率
1999—2014年数据来自文献[13] The data from 1999 to 2014 were extracted from reference [13]
2.3 白叶枯病各近等基因系对测试菌株的抗感性
用954个菌株对含有13个白叶枯病抗性基因的15个近等基因系进行了抗病性测定。抗性频率小于40%的近等基因系有10个,占66.67%;抗性频率大于90%的有4个,分别为IRBB5、IRBB7、CBB23、GDBB23;IRBB21的抗性频率为79.45%(表5)。聚类分析表明,15个水稻白叶枯病近等基因系对954个菌株抗感反应可分为5大类(图3、表6),第Ⅰ类为高感基因系,包括IRBB1、IRBB2、IRBB10、IRBB11、IRBB4;第Ⅱ类为中感基因系,包括IRBB3、IRBB203、IRBB14;第Ⅲ类为中抗基因系,包括IRBB8、IRBB13;第Ⅳ类为抗病基因系,只有IRBB21;第Ⅴ类为高抗基因系,包括IRBB5、IRBB7、CBB23、GDBB23。由此可见,全部用这些近等基因系进行监测会存在冗余性,在建立更精确的鉴别体系时,应去除部分极感品系以及在抗感反应相似度高的品系里抽取出代表品系作为鉴别寄主。
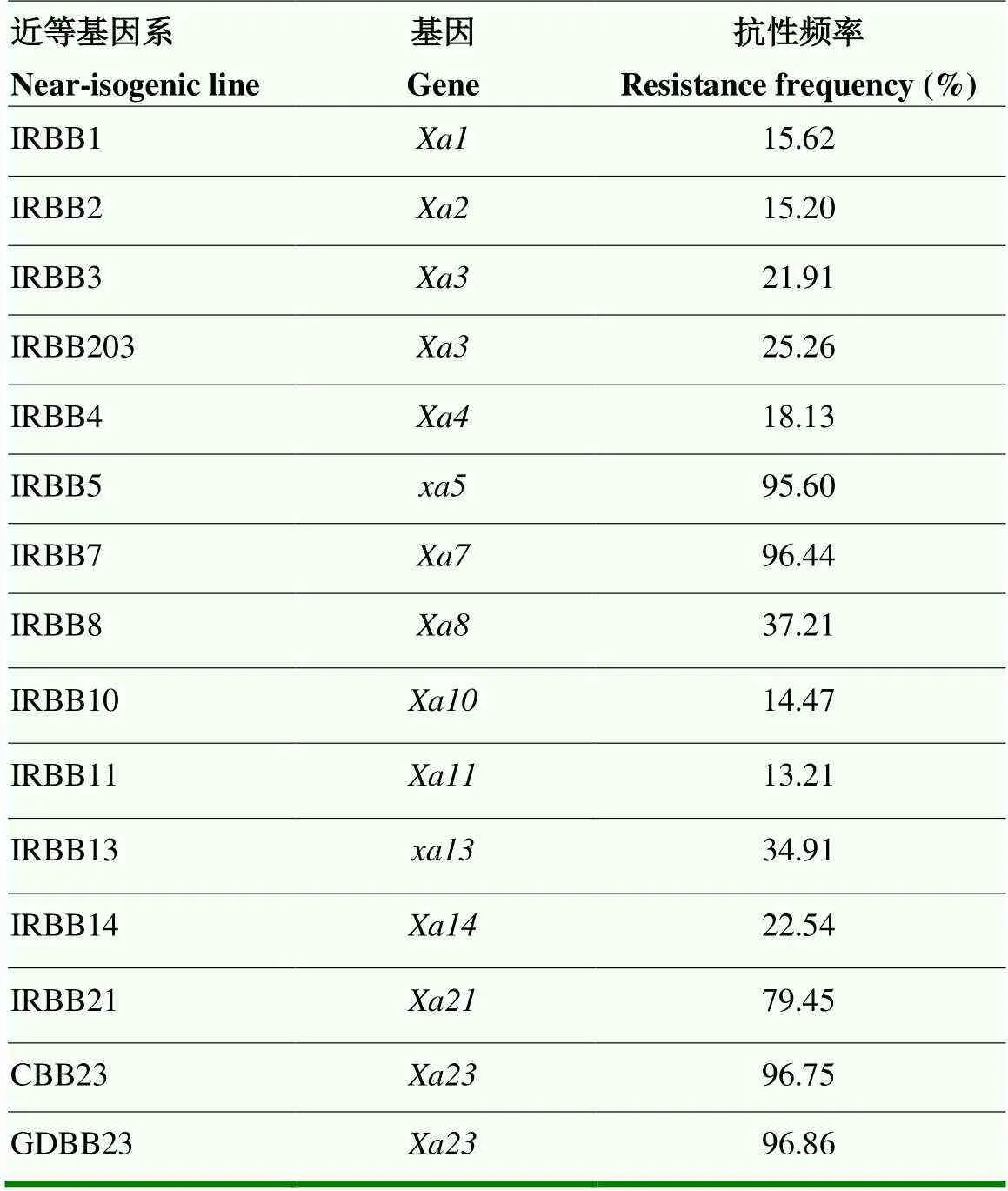
表5 15个水稻白叶枯病近等基因系对954个菌株的抗性频率
在测试的954个菌株中,可侵染广谱抗病基因(IRBB5)的有42个、(IRBB7)的有34个、(CBB23)的有31个。31个可侵染CBB23的菌株有4个可侵染IRBB5、有2个可侵染IRBB7、有6个可侵染IRBB21(表7),可见白叶枯病菌的致病性多样、毒性分化明显,亟需建立鉴别力强的鉴别体系对其进行精准监测。
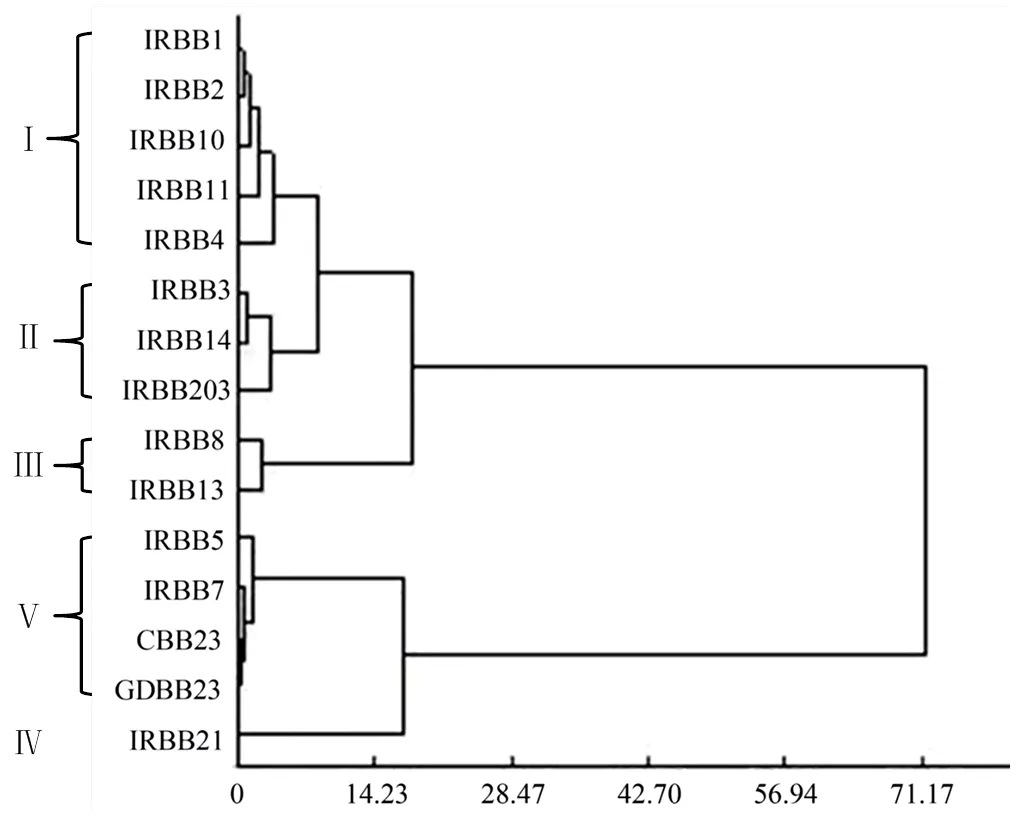
图3 15个水稻白叶枯病近等基因系对954个菌株抗感聚类分析

表6 不同抗性类型的近等基因系对954个白叶枯病菌菌株的抗性频率
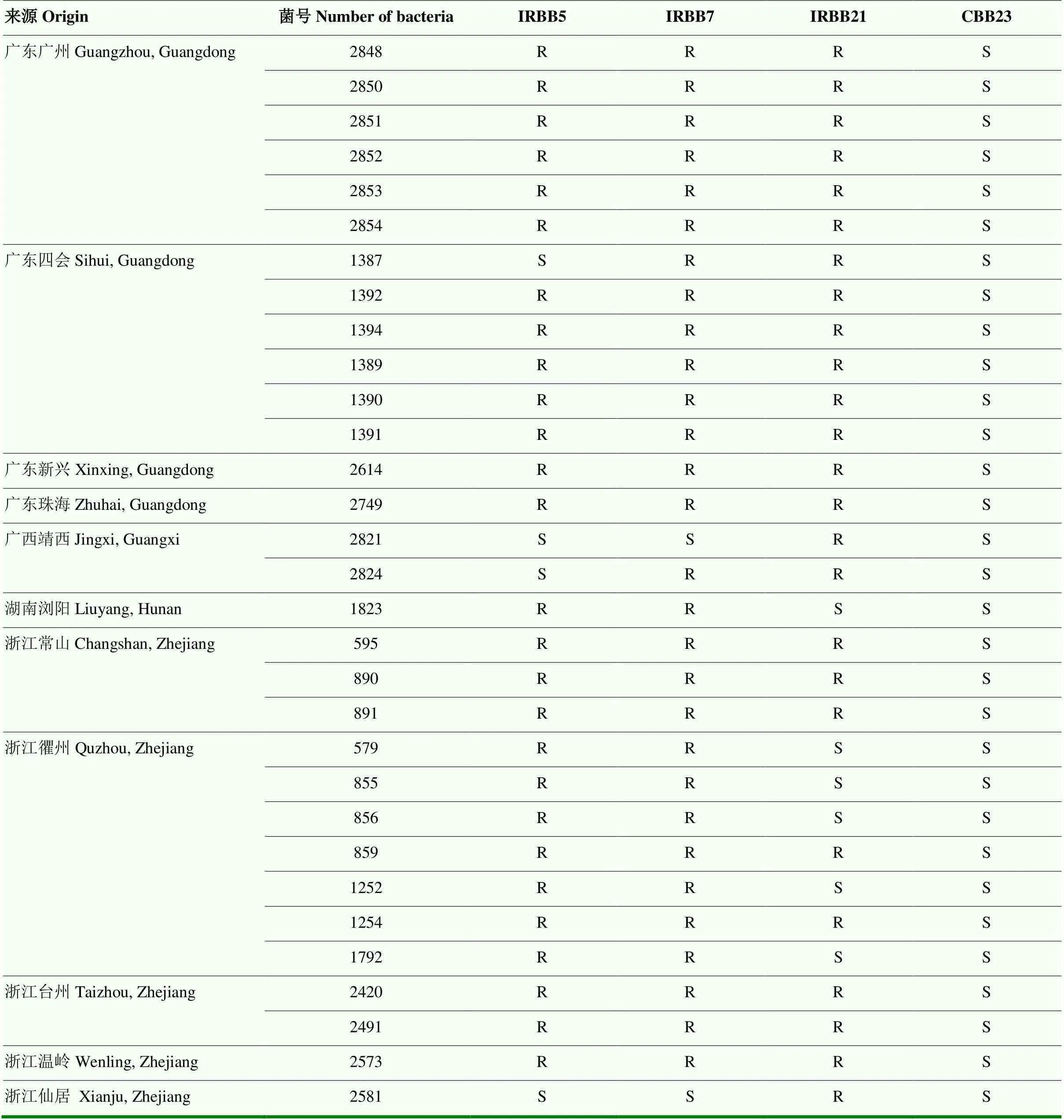
表7 可侵染Xa23菌株对白叶枯病广谱抗性基因的毒性
2.4 中国鉴别寄主鉴定的致病型分化情况
按照应用中国鉴别寄主鉴定的各致病型菌株在15个白叶枯病近等基因系上的抗感反应重新进行归类(表8),发现各致病型在15个白叶枯病近等基因系上均能分化出很多不同的致病型,Ⅰ型、Ⅱ型、Ⅲ型、Ⅳ型、Ⅴ型、Ⅵ型、Ⅸ型、新型菌分别能分化出35、37、44、33、21、6、51、9个致病型。可见目前的中国鉴别寄主并不能很好反应菌株致病性及其无毒因子与白叶枯病抗病基因的互作关系,重新建立新的鉴别体系十分必要。
2.5 近等基因系与病菌互作变量因子分析及新鉴别寄主构建
以15个白叶枯病近等基因系加基因系轮回亲本IR24共16个品种与954个菌株互作的抗感类型为分析对象,感病反应为1,抗性反应为0,组成互作变量数据矩阵,进行变量因子分析。用主成分分析法,以每个近等基因系为变量进行因子提取,且为更好地进行因子变量提取,对因子载荷矩阵进行了方差极大法旋转分析,同时对数据变量矩阵进行Kaiser-Meyer- Olkin(KMO)检验和Bartlett球度检验[24]。分析表明,所组成的数据矩阵KMO值为0.888(KMO值>0.7时,适合因子分析),Bartlett球度检验卡方为10 629.641,相伴概率为0(小于0.05时,适合因子分析),进一步说明该数据矩阵适合因子分析,从中浓缩因子是可行的。
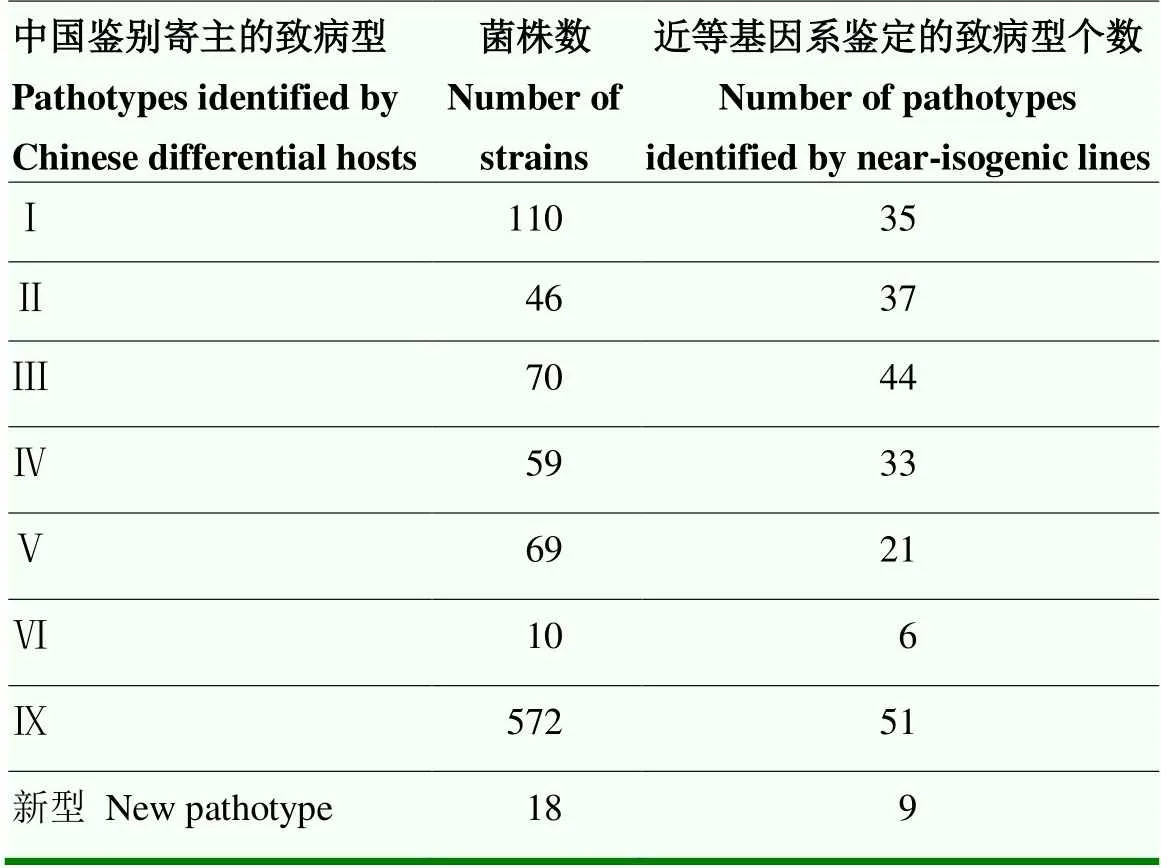
表8 中国鉴别寄主鉴定的各致病型菌株分化情况
对组成的数据矩阵进行主成分因子分析,因子载荷矩阵未旋转前提取了16个因子,解释总方差变量为100%,经方差极大法旋转后提取因子8个,解释变量总方差85.815%(表9)。
依据表10数据,抽提出与8个因子关系密切、载荷系数最高的单基因系。第一因子载荷系数最高的是IRBB10();第二因子载荷系数最高的是CBB23(GDBB23,),第二因子中CBB23与GDBB23因子载荷系数非常相近,CBB23是以金刚30为轮回亲本的粳稻近等基因系,而GDBB23是以CBB23为供体,IR24为受体及轮回亲本的近等基因系,两者所含基因相同,抗性频率也基本一致,但在田间种植时CBB23偏矮、早熟,株型和熟期均不太理想,所以认为选择GDBB23更为理想;第三因子载荷系数最高的是IRBB7(),而IRBB5()跟其位于同一因子中,两者因子载荷系数也相近,但结合2.2中各近等基因系的致病特征(目前已出现可使两者各自感病的菌株)和两者的抗性机制不同,因此可考虑将两者均列入鉴别寄主中;第四因子载荷系数最高的是IRBB21();第五、六因子载荷系数最高的均为IR24();第七因子载荷系数最高的是IRBB13();第八因子载荷系数最高的是IRBB3()。同时,结合各因子抽提的品种中没有一个像CBBD1(金刚30)可被全部测试菌株侵染致感病的品种,考虑将其纳入鉴别寄主中;另外,考虑到位于第一因子的(IRBB4)基因因子载荷系数也较高及其在我国籼稻杂交稻中使用的普遍性,建议将其也纳入鉴别寄主中。因此,综合以上因素和各近等基因系对变量方差贡献大小,初步筛选出IRBB10()、IRBB4()、GDBB23()、IRBB5()、IRBB7()、IRBB21()、IR24()、IRBB13()、IRBB3()、CBBD1(金刚30)共10个品种作为鉴别白叶枯病菌致病型的鉴别寄主。
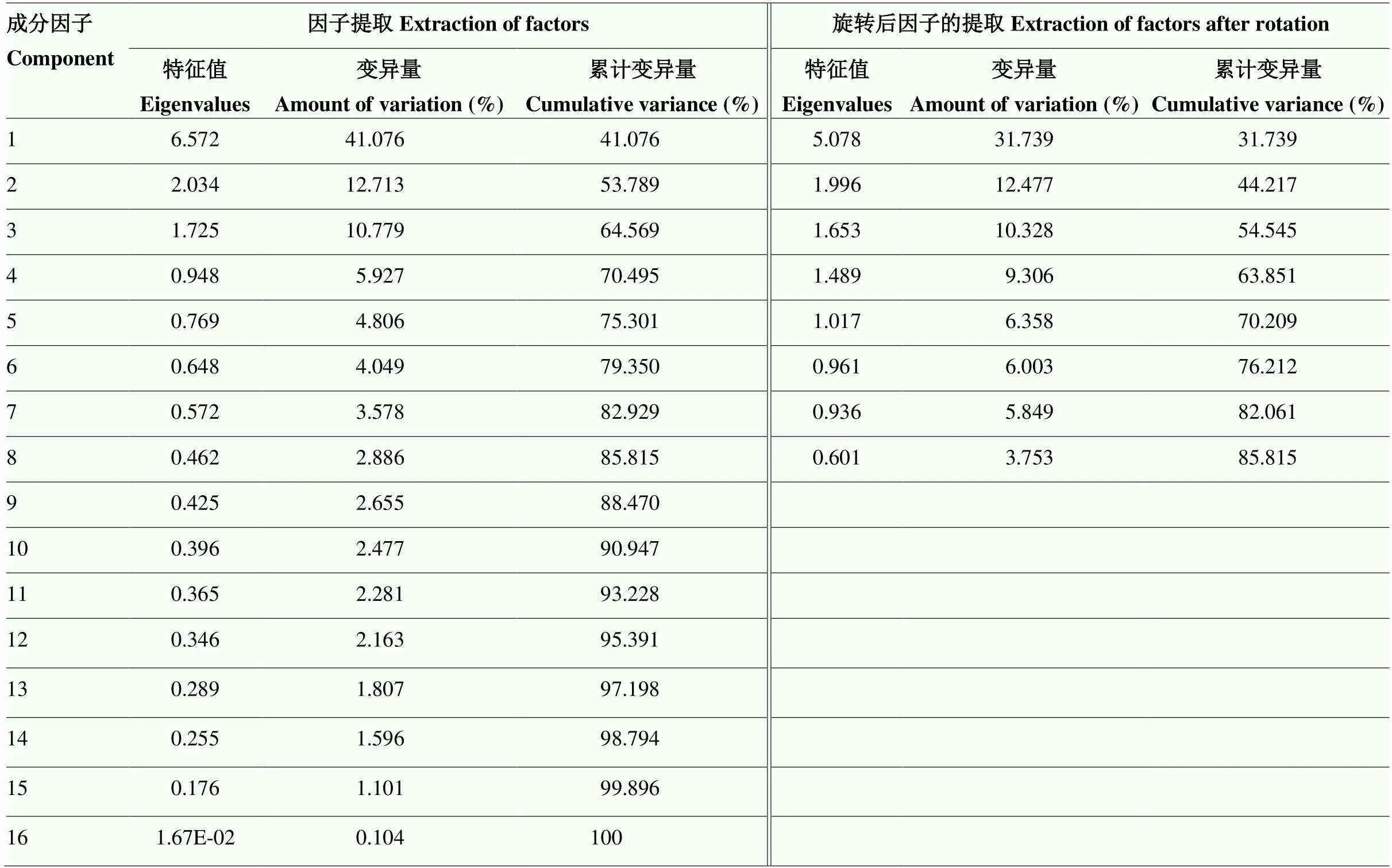
表9 16个近等基因系与954个白叶枯病菌菌株互作的总变异及其因子解释
提取方法Extraction method:主成分分析法 Principal component analysis
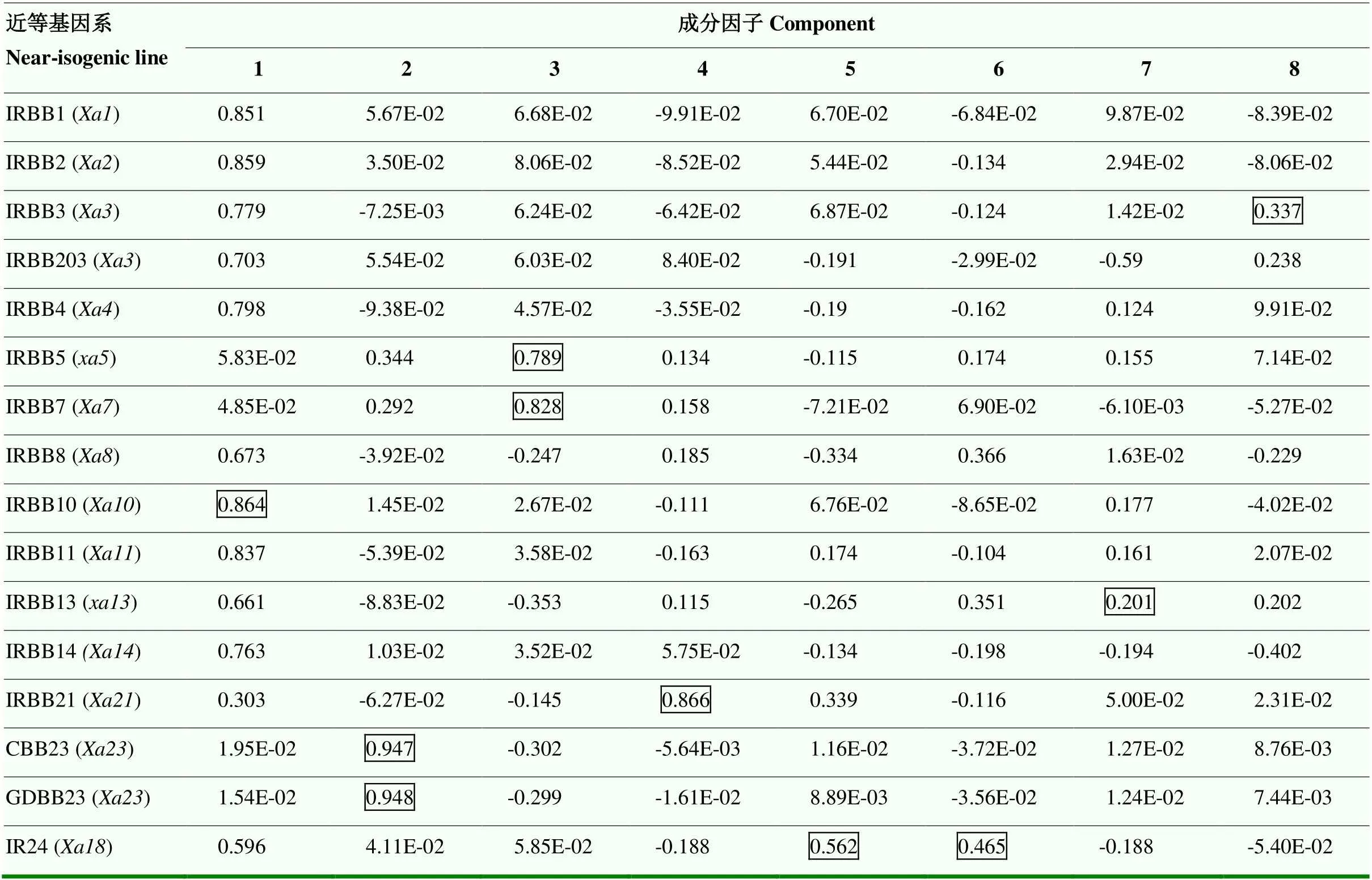
表10 16个近等基因系与954个白叶枯病菌菌株互作的旋转分量矩阵
2.6 新鉴别寄主体系对新小种的划分及其特点
2.6.1 测试菌株对新鉴别系统致病谱特点 根据954个测试菌株在GDBB23、IRBB7、IRBB5、IRBB21、IRBB13、IRBB3、IRBB4、IRBB10、IR24、CBBD1共10个新鉴别寄主上的抗感反应归类,鉴定出55个致病型,暂定为R1—R55(表11)。954个测试菌株中使10个新鉴别寄主致病谱在40%以下的菌株有198个,占20.75%;致病谱在41%—59%的菌株有550个,占57.65%;致病谱在60%以上的菌株有206个,占21.59%。说明测试的白叶枯菌株中,对于10个新鉴别寄主(其中8个为单基因系),中等致病谱的菌株居多,窄致病谱菌株和广致病谱菌株少。其中R32(RRRRSSSSSS)为目前测试地区的优势致病型,有370个,占38.78%;其次是R44(RRRSSSSSSS),有148个,占15.51%。
2.6.2 新鉴别寄主体系与国内其他鉴别体系的鉴别力比较 比较新建立的鉴别寄主体系与国内学者公布的其他5套鉴别体系对测试的954个水稻白叶枯病菌菌株的致病型(表12)以及测试的954个水稻白叶枯病菌菌株对各套鉴别体系的致病谱频数分布(图4),结果表明,本研究新鉴别寄主、陈功友等建立的鉴别寄主[4]、夏立琼等建立的鉴别寄主[22]、杨万风等建立的鉴别寄主[21]、许志刚等建立的鉴别寄主[20]、中国鉴别寄主鉴定出的致病型分别为55、45、39、33、23、11个,以本研究新鉴别寄主鉴定出的致病型最多,而中国鉴别寄主鉴定出的致病型最少。表明前三者对本研究菌株致病性区分更为细化,可分出更多的小种,鉴别力更强。从测试的954个水稻白叶枯病菌菌株对各套鉴别体系的致病谱频数分布图来看,鉴别力较好的本研究新鉴别寄主、陈功友等建立的鉴别寄主、夏立琼等建立的鉴别寄主,致病谱频数分布图的中间值分别为50.4%、43.8%、49.5%,经单样本K-S检验,与正态分布形式比较,这3套鉴别体系中本研究新鉴别寄主、夏立琼等建立的鉴别寄主更符合正态分布。由此说明,基于本研究新鉴别寄主、夏立琼等建立的鉴别寄主鉴别出的病菌种群的致病性趋于正态分布。另外,本文2.2的结果也表明,目前出现可侵染IRBB7()的菌株,而夏立琼等建立的鉴别体系虽然目前对菌株鉴别力也较好,但其缺少了IRBB7();相对于陈功友等建立的鉴别寄主,本研究构建的新鉴别寄主体系主要加入了比IR24更普遍感病的金刚30、用遗传背景为IR24的GDBB23替换了以金刚30为背景的CBB23[17],使得建立的鉴别寄主遗传背景更为一致、加入了与其他近等基因系抗谱有差异的IRBB10,可较好地反映取样稻区菌株毒性的分化及多样性。
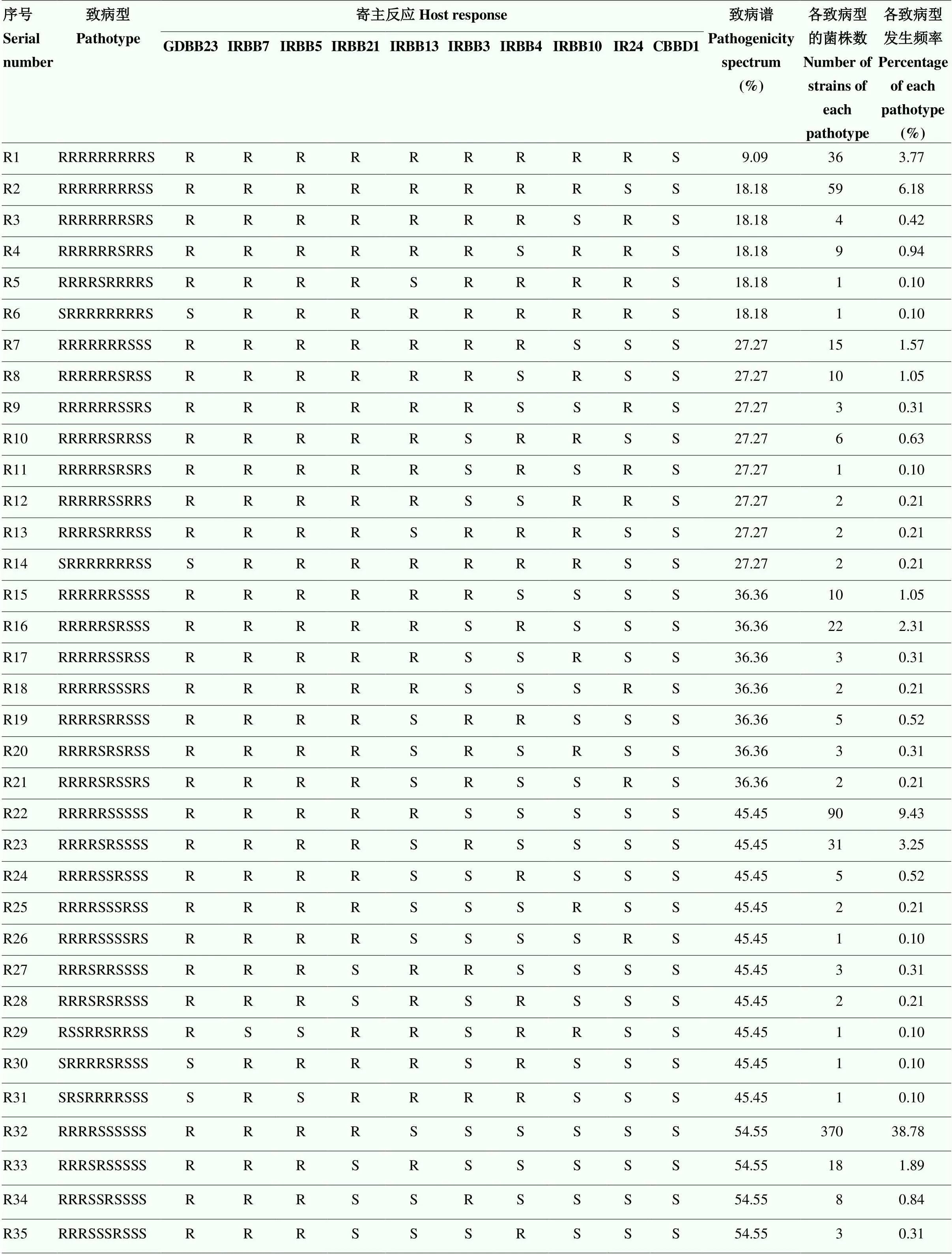
表11 测试菌株在新鉴别寄主上的致病反应
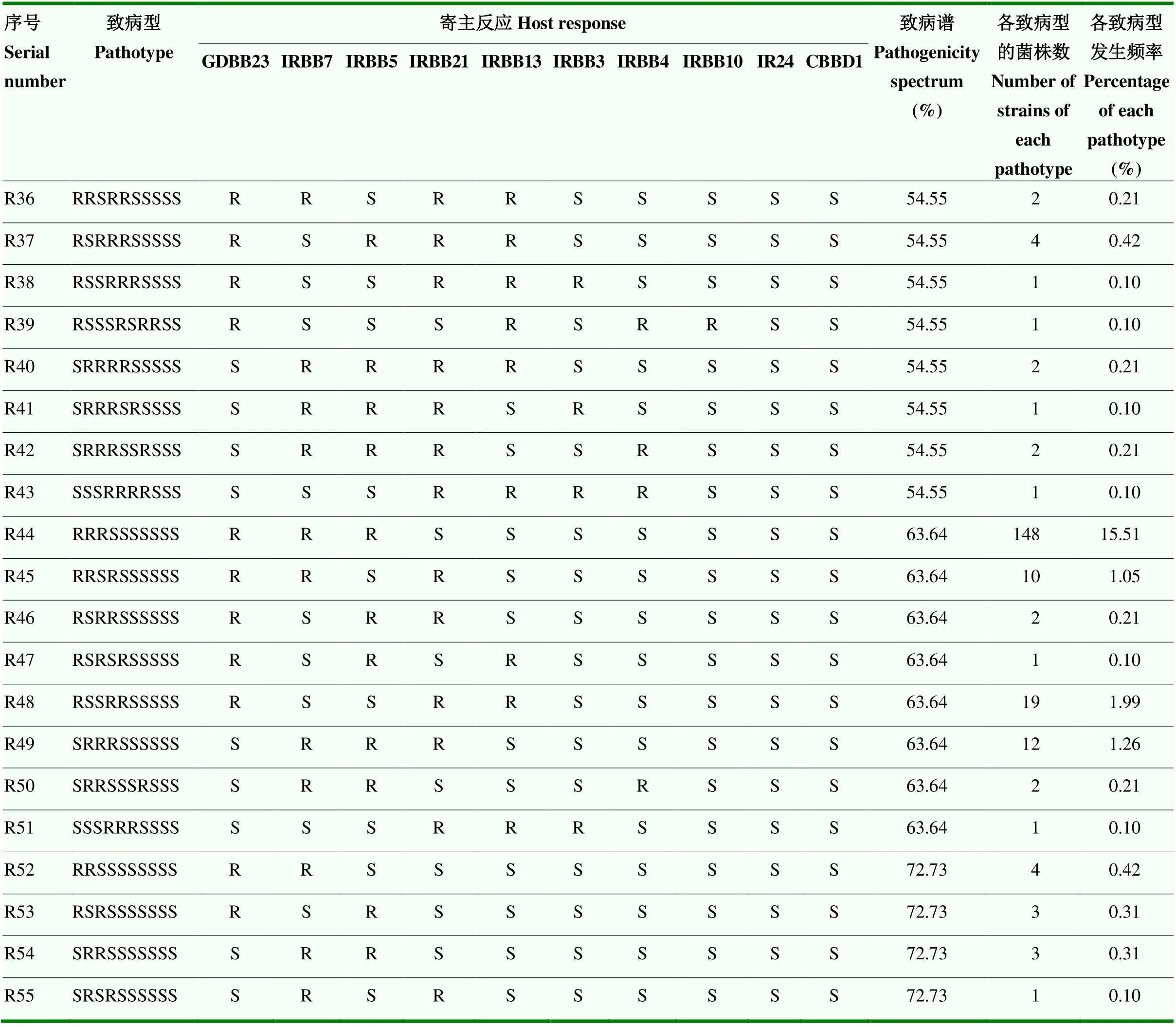
续表11 Continued table 11
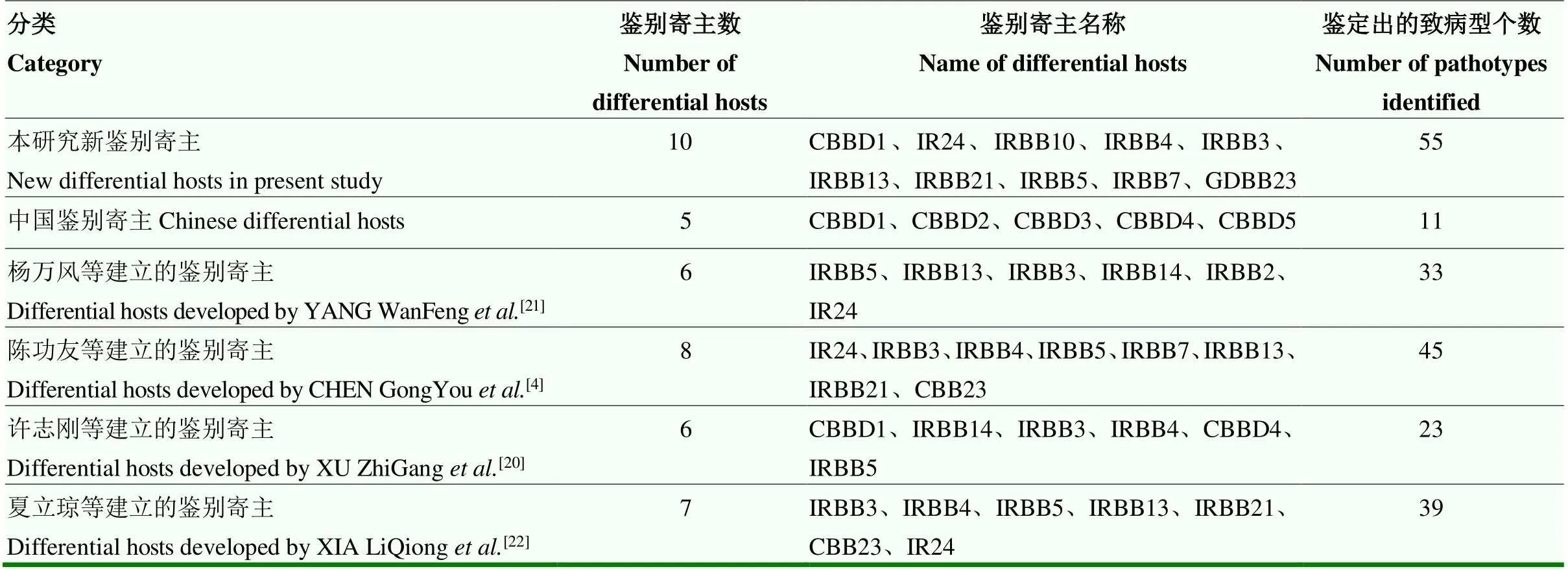
表12 新鉴别寄主体系与国内其他鉴别体系对954个水稻白叶枯病菌菌株的致病型比较
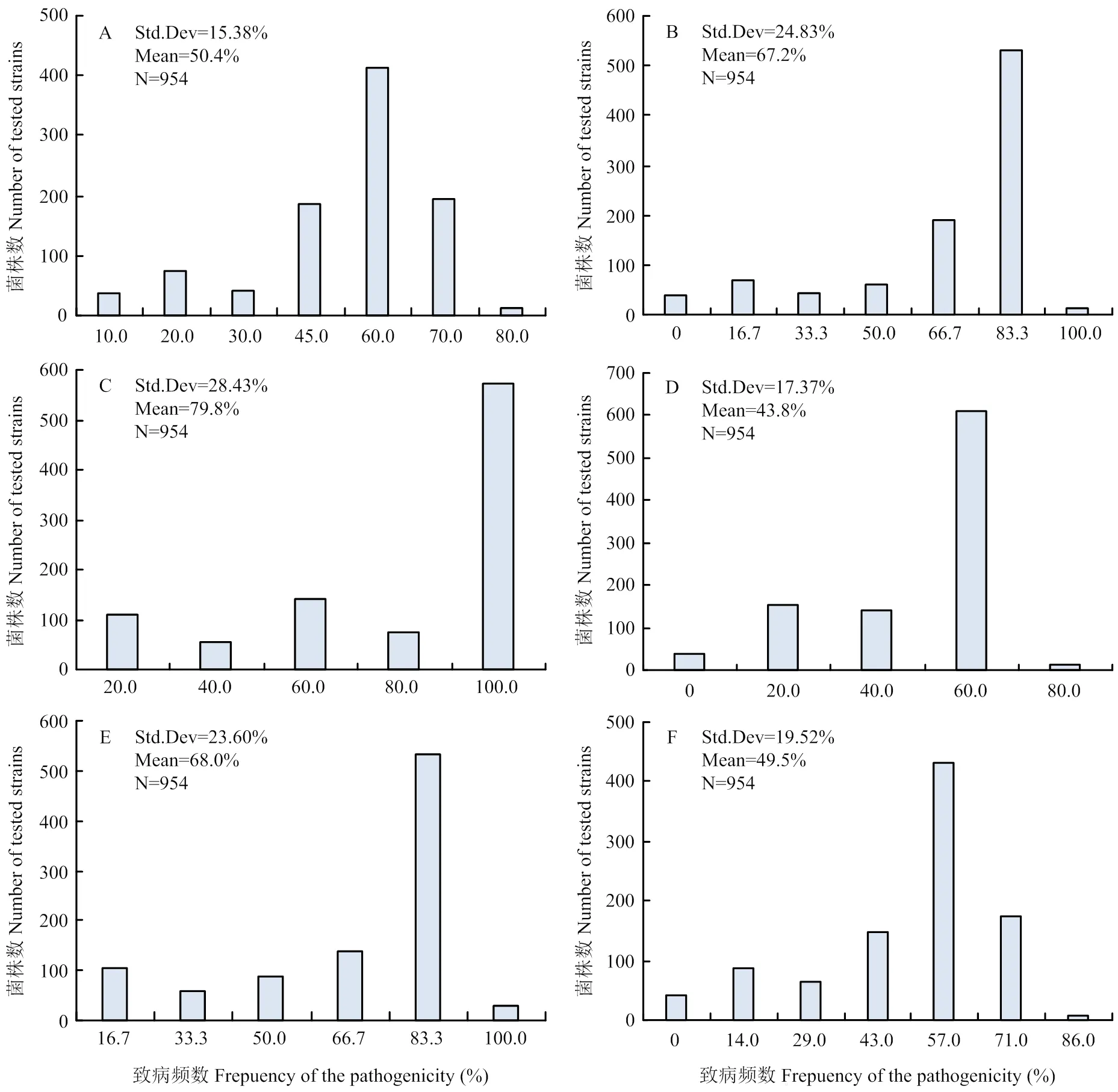
A:本研究新鉴别寄主New differential hosts in present study;B:杨万风等建立的鉴别寄主Differential hosts developed by Yang WanFeng et al.[21];C:中国鉴别寄主Chinese differential hosts;D:陈功友等建立的鉴别寄主Differential hosts developed by Chen Gongyou et al.[4];E:许志刚等建立的鉴别寄主Differential hosts developed by Xu ZhiGang et al.[20];F:夏立琼等建立的鉴别寄主Differentials developed by Xia LiQiong et al.[22]
2.7 白叶枯病抗病基因聚合效应预测
不同白叶枯病抗性基因对954个菌株的抗感聚合分析结果表明,2、3、4或5个基因聚合预测均提升了抗性频率,抗病基因之间对测试菌株的抗性有一定的互补性(表13)。2个基因聚合中,分别与聚合效应比与其他抗病基因的好,聚合后抗性频率分别提高了3.88%、2.94%、2.63%、1.79%,均能使抗性频率提高到80%以上;相互聚合或分别与聚合效应较好,均能使抗性频率提高到40%以上,与聚合,抗性频率提高了8.18%,达到45.39%;高抗基因互相聚合或与聚合后亦能使抗性频率有所提高,与聚合效应最好,聚合后抗性频率达到了99.79%。3个基因聚合中,、分别与与聚合后效应最好,聚合后抗性频率分别提高了9.23%、11.22%、11.53%,抗性频率分别达到46.44%、48.43%、48.74%;与中抗、感病或中感基因、高抗基因三基因聚合后,都能使抗性频率提高到80%以上。4个基因聚合中,与聚合效应较好,聚合后抗性频率分别提高了9.23%、12.37%,抗性频率分别达到46.44%、49.58%;聚合后抗性频率达到了99.58%。5个基因聚合中,聚合后抗性频率提高了5.04%,抗性频率为84.49%;聚合后抗性频率达到了100%。
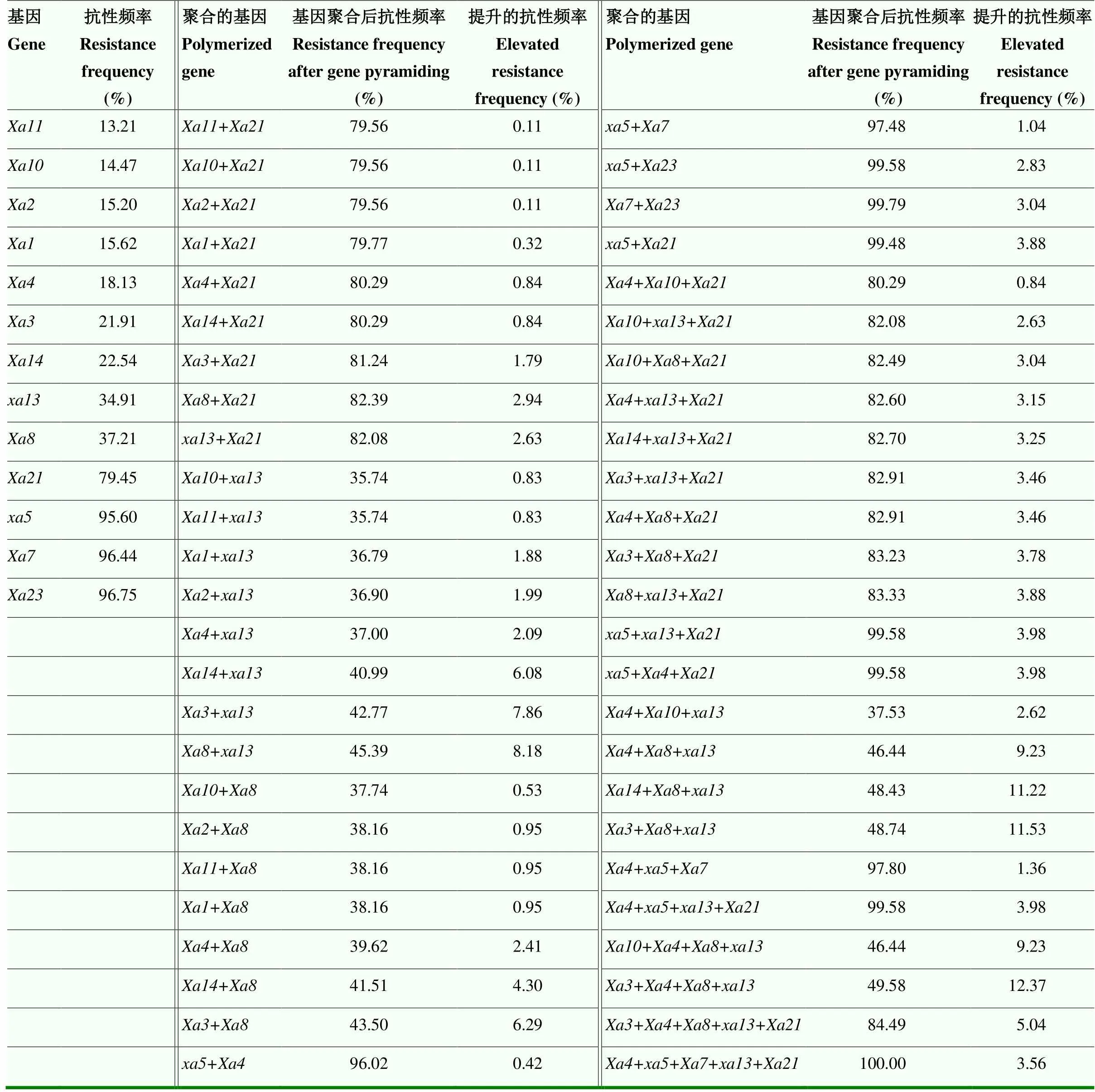
表13 白叶枯病抗病基因聚合效应
3 讨论
3.1 水稻白叶枯病菌毒性分化
我国经历了从20世纪80—90年代广泛种植含有的品种,到近年来种植含等单基因或聚合基因的品种[25-28]。水稻品种所含抗性基因的更新迭代以及水稻生态系统和气候环境变化加速了病原菌的致病性演变。白叶枯病菌的TALE基因和水稻的R基因长期相互选择,表现为军备竞赛状态[4],因此,白叶枯病菌毒性出现分化、各地优势致病型不断变化是必然的。本研究系统报道了监测地区白叶枯病菌的致病性,通过利用中国鉴别寄主、国内外构建的抗白叶枯病近等基因系共21个鉴别品种从2018—2021年监测了我国7个水稻生产大省954个单菌落菌株的致病性,结果表明监测地区白叶枯病菌趋向多样化,毒性分化明显。本研究表明监测地区强毒菌系Ⅸ型菌已上升为优势致病型,与陈深等[13]报道的华南稻区以V型菌为优势致病型相比发生了变化,说明该地区的白叶枯病菌正向毒性更强的方向发展。在本研究测试菌株中,出现可侵染的有42个、的有34个、的有31个、的有196个,的有781个,这些菌株对广谱抗病基因抗感反应均有差异,如侵染的菌株不一定都能侵染或,表明测试稻区白叶枯病菌的致病性多样、毒性分化明显。水稻白叶枯病广谱抗性基因被克服在前人的研究中有相继报道,2006年,杨万风等[21]研究表明采自云南的1个菌株YN24能对IRBB5()高度致病;2015年,陈小林等[29]报道5个来自广西南宁的菌株可使CBB23感病;2018年,袁斌等[30]在测试湖北省13个白叶枯病菌菌株时发现有2个菌株可使IRBB7()致病;2019年,陈功友等[4]报道在500余株白叶枯病菌菌株中也发现广谱抗病基因和可分别被6、11、2个菌株克服,的抗性可被多数菌株克服,而抗性可被82.4%的测试菌株克服。以上研究揭示我国白叶枯病菌均致病性多样,优势菌系正在不断演变,这是我国部分稻区近年白叶枯病暴发成灾的重要原因之一。为此,加强白叶枯病菌毒性变异监测、加快新抗病基因的挖掘、广谱抗病品种创制以及新型抗病品种的选育应用是实现白叶枯病可持续防控的重要途径及措施。
3.2 水稻白叶枯病菌致病型监测鉴别体系构建
对白叶枯病菌毒性变化的精准监测,既可为抗病育种提供准确的病菌小种靶标,又可对生产上的品种开展抗性监测预警, 指导品种的布局与轮换,对白叶枯病的防控意义重大。一直以来,病原菌的生理小种(致病型)主要以菌株在鉴别寄主上的一系列抗性反应型进行划分,这种方法比较直观、可靠,具有较好的生物学意义,但鉴别寄主的选择直接影响并反映菌株毒性分化的精准性。本研究同时采用中国鉴别寄主和含有13个白叶枯病抗性基因的15个近等基因系两套鉴别寄主,对监测地区的白叶枯病菌致病性分化进行了测定。结果表明中国鉴别寄主虽然能适合本土气候、病健易于识别,品种间的抗感性梯度差异亦可对低毒和中毒菌株具有较好的鉴别能力,但一方面本土菌株已出现较多强致病菌系(Ⅸ型菌)致所有鉴别寄主抗性被克服,另一方面大部分鉴别品种遗传背景不清楚,不能准确反映致病型与抗性基因的互作关系,本研究通过对比发现用其鉴定的各个致病型在15个白叶枯病近等基因系上均能分化出很多不同的致病型,可见其局限性已非常明显,而应用已知抗病基因的近等基因系可弥补其局限性,因此,筛选应用已知抗病近等基因系进行白叶枯病菌的致病性监测已经成为趋势。许志刚等[20]选用金刚30、IRBB14、IRBB3、IRBB4、Java14和IRBB5对来自不同地区的100株菌株进行鉴定,将中国的白叶枯病菌区分为8个小种(C1—C8);杨万风等[21]选用IRBB5、IRBB13、IRBB3、IRBB14、IRBB2和IR24对来自1970—1992年、2003—2004年全国不同地区的285个菌株进行鉴定,分为R1—R9共9个生理小种;陈深等[13]选用与杨万风相同的鉴别寄主对来自华南地区不同生态稻区的150个菌株进行鉴定,分为R1、R2、R3、R4、R5、R8、R10 7个致病小种,其中R8为优势小种;夏立琼等[22]选用IRBB3、IRBB4、IRBB5、IRBB13、IRBB21、CBB23和IR24对来自海南18个市的25个菌株进行鉴定,分出6个致病型;陈功友等[4]选用IR24、IRBB3、IRBB4、IRBB5、IRBB7、IRBB13、IRBB21、CBB23对10年间在全国采集的500多株菌株进行了鉴定,重点从菌株的tal基因和R基因的互作关系将菌株进行了分类;Kogeethavani等[23]通过利用9个白叶枯病近等基因系将40个马来西亚菌株分成21个致病小种,其中R20为优势小种,可使当地大部分品种感病。
本研究基于测试菌株与15个近等基因系及IR24的抗感互作,应用主成分因子分析法,开展近等基因系与病菌互作的变量因子分析,构建了白叶枯病菌致病型近等基因系鉴别寄主。主成分分析方法(PCA)是一种用较少变量代替原样本中较多变量的线性变换方式,变换后的较少变量被称作新变量,新变量在一定程度上包含原变量较多信息量[31]。PCA主要功能是找出变量的浓缩因子,分析多个变量间的关系,构建变量间的总体性指标,避免变量数过多,造成解释上的复杂与困扰,在尽量不丧失原有信息的前提下,抽取少数几个主成分,作为代表原来变量的总体性指标,达到数据缩减的功能。PCA作为一种统计学方法,目前已广泛应用于医学、化学、电子信息学及生物学领域[32-35]。在水稻全基因组关联作图分析中,PCA应用于性状与基因组变异的互作变量分析[36-37];在全基因组表达谱研究中,PCA应用于复杂而庞大的数据处理,可完成对代谢路径等不同基因网络的归类及相关性分析[38];在植物病害研究方面,主要应用于病害的识别、病害发生预测和鉴别寄主的筛选。在病害的识别研究中,通过PCA对复杂的特征参数降维,以提取主要参数,成功对不同病害形成分类识别技术和高效的分类器[30]。在病害发生预测分析中,通过PCA对植物叶片叶绿素荧光光谱进行降维,提取光谱特征信息,建立精准的病害识别和预警模型[39]。本研究将PCA引入白叶枯病近等基因系鉴别体系的筛选,从鉴别寄主与菌株大量而多样的互作数据中浓缩因子,初步筛选了既能反映寄主与菌株互作关系又能抽提出不同类型的抗病近等基因系,避免建立的鉴别寄主之间的重叠及代表性不足,与国内其他鉴别体系比较,本研究构建的新鉴别体系能较好反映出测试稻区白叶枯病菌小种的多样性。
通过与国内其他[4,20-22]鉴别体系对比,新鉴别寄主体系对本研究菌株致病性区分更为细化,可分出更多的小种,鉴别力较强。本套鉴别寄主有以下特点:首先,基本涵盖了能反应病菌与抗病基因互作的各类代表不同基因结构和分子机制的近等基因系。目前被鉴定的46个白叶枯病主效基因中已经被克隆了18个[40-42],根据其基因结构和分子机制可分为6类:第1类、、、、(t)、(t),编码含有非典型核苷酸结合位点-富含亮氨酸重复序列(NBS-LRR)的蛋白质;第2类和,编码细胞外LRR受体激酶;第3类,编码一种细胞壁相关激酶;第4类,编码一般转录因子的小亚基;第5类编码糖转运蛋白家族的和(t);第6类抗病执行基因和、。本套鉴别寄主10个品种中已含有、、、、、、、共8个已克隆白叶枯抗病基因的近等基因系,代表了上述基因结构和分子机制不同的5大类基因和各级别抗谱品种。没有涵盖第一类基因,一方面是因为这类基因均是极感系统,另一方面已有研究证明(t)、、(t)、均是的等位基因[43-44],而在本研究数据因子提取时已表明(表10),第一因子载荷系数最高的虽是IRBB10(),但也非常相近,三者中任一个均可代表,同时考虑IRBB10比IRBB1、IRBB2更适应本地区气候和株型适合,因此最终选择了IRBB10。其次,本套鉴别寄主以本研究室发展的IR24为背景、含的近等基因系GDBB23替换了CBB23(),使遗传背景、生育期及株型与其他近等基因系保持一致,避免了CBB23在监测地区遗传背景与其他近等基因系不一致、生育期短等问题,此外,加入了对测试的所有菌株普遍感病的高感品种金刚30(CBBD1,中国鉴别寄主),作为感病对照品种。本套新建立的鉴别寄主体系是依据我国部分稻区的白叶枯病菌毒性情况及现有的近等基因系建立起来的,由于我国稻区辽阔、品种结构复杂,因此,该鉴别寄主系统存在着一定的时空局限性,在其他稻区的应用还要根据不同稻区以及不同时期病原结构特点加以甄别、调整及完善,以便更好地服务于指导水稻生产中白叶枯病的预警以及抗病品种布局。
3.3 多基因聚合是培育持久抗性品种的重要途径
前人研究显示白叶枯病抗病基因对白叶枯病菌具有小种特异性[28],抗性被克服的实例表明,单个抗病基因抵御水稻白叶枯病菌的时效性有限,因此,将不同抗谱、不同机制的抗病基因聚合是开发白叶枯病持久抗性品种的重要策略。目前将多个抗病基因聚合到优异感病品种的研究已有报道,其中是应用于聚合较多的基因,实践证明这些基因2、3、4、5个聚合后品种抗性均可增加[28],这与本研究统计预测的结果相似。本研究通过抗感反应聚合的 2、3、4或5个基因,聚合后抗性频率均有一定的提升,说明抗病基因之间对测试菌株的抗性具有互补性,聚合后能产生一定的加性效应。周俊飞等[25,45-46]研究证明将分别聚合到深水稻品种Jalmagna、耐盐水稻品种CSR-30、IRBB59,均取得了良好的抗性;Hsu等[47]聚合了、、、、,育成了中国台湾较受欢迎的粳稻品种台农82。而本研究也证明、、聚合后抗性频率达到99.58%、、、、、聚合后抗性频率达到100%,研究结果可为监测稻区的后续基因聚合抗病育种提供参考。然而,基因聚合也有弱化品种抗性的报道,如与、、等基因聚合的抗性叠加效应不明显[25],因此,基因聚合尚需考虑抗性基因的特点以及受体品种的遗传背景。聚合抗病基因是提高品种抗性以及持久性的有效途径,但同时多基因聚合品种的广泛种植也会对病菌施加巨大的选择压力,驱使病原菌逃避植物免疫识别,加速超级致病菌小种的出现,导致病害出现无法持续有效控制的大发生局面[48]。为此,持续发掘创制新抗病基因资源、发展不同途径的病害控制方法,将有助于白叶枯病的可持续控制。
4 结论
致病性广的强毒菌系Ⅸ型菌已上升为华南稻区(广东、广西、海南)、长江中下游稻区(湖南、浙江)的优势致病型;西南稻区(云南)以Ⅳ型菌为主;东北稻区(辽宁)仍以I型菌为主;监测地区的白叶枯菌系趋向多样化,毒性分化明显,可侵染广谱抗病基因的菌株已出现并有上升趋势;应及时调整抗病育种策略,科学制定抗病品种防控布局措施。以GDBB23()、IRBB7()、IRBB5()、IRBB21()、IRBB13()、IRBB3()、IRBB4()、IRBB10()、IR24()、CBBD1(金刚30)10个鉴别品种组成的白叶枯病菌新鉴别体系,可将监测地区的954个测试菌株划分为55个致病型,新鉴别寄主体系以抗白叶枯病近等基因系为主,能较好地反映菌株与抗病基因的互作、准确监测白叶枯病菌毒性分化。
[1] 方中达. 水稻白叶枯病. 南京: 江苏人民出版社, 1963: 1-2.
FANG Z D. Rice Bacterial Blight. Nanjing: Jiangsu People’s Publishing Press, 1963: 1-2. (in Chinese)
[2] 章琦. 水稻白叶枯病抗性的遗传及改良. 北京: 科学出版社, 2007: 2-4.
ZHANG Q. Genetics and Improvement of Resistance to Bacterial Blight in Rice. Beijing: Science Press, 2007: 2-4. (in Chinese)
[3] LEACH J E, WHITE F F. Bacterial avirulence genes.Annual Review of Phytopathology, 1996, 34: 153-179.
[4] 陈功友, 徐正银, 杨阳阳, 邹丽芳, 朱勃. 我国水稻白叶枯病菌致病型划分和水稻抗病育种中应注意的问题. 上海交通大学学报(农业科学版), 2019, 37(1): 67-73.
CHEN G Y, XU Z Y, YANG Y Y, ZOU L F, ZHU B. Classification of pathotypes of Chinesepv.and resistance breeding strategies for bacterial blight. Journal of Shanghai Jiaotong University (Agricultural Science), 2019, 37(1): 67-73.(in Chinese)
[5] 堀野修, 季伯衡. 水稻白叶枯病菌的小种分化与水平抗性鉴定法. 农业译文, 1991(2): 1-7.
HORINO O, JI B H.Race differentiation ofpv.and horizontal resistance identification.Agricultural Translation, 1991(2): 1-7. (in Chinese)
[6] NODA T, HORINO O, OHUCHI A. A survey of geographical distribution of pathogenic races ofpv.from Japan. Proceedings of the Association for Plant Protection of Hokuriku, 1987, 35: 7-13. (in Japanese with English summary)
[7] NODA T, OHUCHI A. A new pathogenic race ofpv.and inheritance of resistance of differential rice variety, Te-tep to it. Japanese Journal of Phytopathology, 1989, 55(2): 201-207.
[8] MEW T W. Current status and future prospects of research on bacterial blight of rice.Annual Review of Phytopathology, 1987, 25: 359-382.
[9] 伍尚忠, 徐羡明, 刘景梅, 苗东华, 维拉·克鲁茨. 华南及菲律宾稻白叶枯病病原菌株致病性比较研究. 植物病理学报, 1985, 15(2): 65-72.
WU S Z, XU X M, LIU J M, MEW T W, VERA CRUZ C M.Comparison of virulence ofpv.in south
china and in the philippines. Acta Phytopathologica Sinica, 1985, 15(2): 65-72. (in Chinese)
[10] 方中达, 许志刚, 过崇俭, 殷尚智, 伍尚忠, 徐羡明, 章琦. 中国水稻白叶枯病菌致病型的研究. 植物病理学报, 1990, 20(2): 81-88.
FANG Z D, XU Z G, GUO C J, YIN S Z, WU S Z, XU X M, ZHANG Q. Studies on pathotypes ofpv.in china. Acta Phytopathologica Sinica, 1990, 20(2): 81-88. (in Chinese)
[11] 王春连, 章琦, 周永力, 赵炳宇. 我国长江以南地区水稻白叶枯病原菌遗传多样性分析. 中国水稻科学, 2001, 15(2): 131-136.
WANG C L, ZHANG Q, ZHOU Y L, ZHAO B Y.Genetic diversity of pathogenpv.from southern regions of Yangtze River in china.Chinese Journal of Rice Science, 2001, 15(2): 131-136. (in Chinese)
[12] 曾列先, 朱小源, 杨健源, 伍圣远, 陈珍, 陈深. 广东水稻白叶枯病菌新致病型的发现及致病性测定. 广东农业科学, 2005(2): 58-59.
ZENG L X, ZHU X Y, YANG J Y, WU S Y, CHEN Z, CHEN S.A new pathotype ofpv.was found and tested for pathogenicity in Guangdong. Guangdong Agricultural Sciences, 2005(2): 58-59. (in Chinese)
[13] 陈深, 汪聪颖, 苏菁, 冯爱卿, 朱小源, 曾列先. 华南水稻白叶枯病菌致病性分化检测与分析. 植物保护学报, 2017, 44(2): 217-222.
CHEN S, WANG C Y, SU J, FENG A Q, ZHU X Y, ZENG L X. Differential detection and analysis of pathotypes and differentiation againstpv.in southern China. Journal of Plant Protection, 2017, 44(2): 217-222. (in Chinese)
[14] OGAWA T, KHUSH G S. Major genes for resistance to bacterial blight in rice//Bacterial Blight of Rice. Internationa Rice Research Institute, 1989.
[15] OGAWA T, YAMAMOTO T, KHUSH G S, MEW T W. Breeding of near-isogenic lines of rice with single genes for resistance to bacterial blight pathogen (pv.). Japanese Journal of Breeding, 1991, 41(3): 523-529.
[16] 章琦, 杨文才, 施爱农, 王春莲, 阙更生, 赵炳宇, 邢全党. 3个粳稻抗白叶枯病近等基因系的构建. 作物学报, 1998, 24(6): 799-804.
ZHANG Q, YANG W C, SHI A N, WANG C L, QUE G S, ZHAO B Y, XING Q D. Breeding of three near-isogenic japonica rice lines with major genes for resistance to bacterial-blight. Acta Agronomica Sinica, 1998, 24(6): 799-804. (in Chinese)
[17] 章琦, 王春连, 赵开军, 杨文才, 乔枫, 周永力, 江祺祥, 刘古春. 携有抗白叶枯病新基因水稻近等基因系的构建及应用. 中国水稻科学, 2002, 16(3): 206-210.
ZHANG Q, WANG C L, ZHAO K J, YANG W C, QIAO F, ZHOU Y L, JIANG Q X, LIU G C. Development of near-isogenic line CBB23 with a new resistance gene to bacterial blight in rice and its application. Chinese Journal of Rice Science, 2002, 16(3): 206-210. (in Chinese)
[18] JIANG N, YAN J, LIANG Y, SHI Y, HE Z, WU Y, ZENG Q, LIU X, PENG J. Resistance genes and their interactions with bacterial blight/leaf streak pathogens () in rice (L.)—an updated review. Rice, 2020, 13(1): 3.
[19] NEELAM K, MAHAJAN R, GUPTA V, BHATIA D, GILL B K, KOMAL R, LORE J S, MANGAT G S, SINGH K. High-resolution genetic mapping of a novel bacterial glight resistance gene-identified fromand transferred to. Theoretical and Applied Genetics, 2020, 133(3): 689-705.
[20] 许志刚, 孙启明, 刘凤权, 陈志谊, 胡白石, 郭亚辉, 刘永峰, 刘红霞. 水稻白叶枯病菌小种分化的监测. 中国水稻科学, 2004, 18(5): 469-472.
XU Z G, SUN Q M, LIU F Q, CHEN Z Y, HU B S, GUO Y H, LIU Y F, LIU H X. Race monitoring of rice bacterial blight (pv.) in china. Chinese Journal of Rice Science, 2004, 18(5): 469-472. (in Chinese)
[21] 杨万风, 刘红霞, 胡白石, 许志刚, 刘凤权. 中国水稻白叶枯病菌毒性变异研究. 植物病理学报, 2006, 36(3): 244-248.
YANG W F, LIU H X, HU B S, XU Z G, LIU F Q. Virulence variation ofpv.on rice near-isogenic lines in China. Acta Phytopathologica Sinica, 2006, 36(3): 244-248. (in Chinese)
[22] 夏立琼, 李明容, 谢仕猛, 翟文学, 夏志辉. 海南水稻白叶枯病菌优势生理小种的分离及致病力分析. 分子植物育种, 2016, 14(5): 1336-1340.
XIA L Q, LI M R, XIE S M, ZHAI W X, XIA Z H. Isolation and pathogenicity analysis of the predominant race ofpv.in hainan. Molecular Plant Breeding, 2016, 14(5): 1336-1340. (in Chinese)
[23] KOGEETHAVANI R, FATIN N A, SUZIANTI I V, ERWAN S S. Characterization of pathogenic variability ofpv.isolates causing bacterial leaf blight disease in Malaysian rice granaries. Australasian Plant Pathology, 2021, 50: 293-298.
[24] 余建英, 何旭宏. 数据统计分析与SPSS应用. 北京: 人民邮电出版社, 2003.
YU J Y, HE X H. Data statistical analysis and spssapplication. Beijing: People’s Posts and Telecommunications Press, 2003. (in Chinese)
[25] 周俊飞, 章山, 孙婧, 张伟, 高利芬. 水稻抗白叶枯病近等基因系的抗性鉴定揭示基因聚合的多向性效应. 华北农学报, 2020, 35(6): 67-73.
ZHOU J F, ZHANG S, SUN J, ZHANG W, GAO L F. Resistance identification of near-isogenic lines to bacterial blight in rice reveals the multidirectional effect of gene pyramiding. Acta Agriculture Boreali-Sinica, 2020, 35(6): 67-73. (in Chinese)
[26] 成太辉, 陈深, 杨健源, 朱小源, 伍圣远, 洪启金, 曾列先. 水稻抗白叶枯病V型菌基因利用现状及前景. 广东农业科学, 2020, 47(1): 92-97.
CHENG T H, CHEN S, YANG J Y, ZHU X Y, WU S Y, HONG Q J, ZENG L X. Dtilization situation and prospect of geneagainst pathotype v of rice bacterial blight. Guangdong Agricultural Sciences, 2020, 47(1): 92-97. (in Chinese)
[27] 倪大虎, 易成新, 杨剑波, 汪秀峰, 张毅, 章琦, 王春连, 赵开军, 王文相, 李莉. 利用分子标记辅助选择聚合(t)和基因. 分子植物育种, 2007, 5(4): 491-496.
NI D H, YI C X, YANG J B, WANG X F, ZHANG Y, ZHANG Q, WANG C L, ZHAO K J, WANG W X, LI L. Pyramiding(t) andgenes by molecular marker-assisted selection. Molecular Plant Breeding, 2007, 5(4): 491-496. (in Chinese)
[28] ABDUL FIYAZ R, SHIVANI D, CHAITHANYA K, MOUNIKA K, CHIRANJEEVI M, LAHA G S, VIRAKTAMATH B C, SUBBA RAO L V, SUNDARAM R M. Genetic improvement of rice for bacterial blight resistance: present status and future prospects. Rice Science, 2022, 29(2): 118-132.
[29] 陈小林, 颜群, 高利军, 韦善富, 李道远, 高汉亮. 广西水稻白叶枯病菌致病型的初步鉴定. 南方农业学报, 2015, 46(2): 236-240.
CHEN X L, YAN Q, GAO L J, WEI S F, LI D Y, GAO H L. Preliminary identification of pathotype ofpv.in Guangxi. Journal of Southern Agriculture, 2015, 46(2): 236-240. (in Chinese)
[30] 袁斌, 刘友梅, 黄薇, 张舒, 吕亮, 常向前, 杨小林. 湖北省水稻白叶枯病菌致病型分化检测与分析. 湖北农业科学, 2018, 57(24): 100-103.
YUAN B, LIU Y M, HUANG W, ZHANG S, LÜ L, CHANG X Q, YANG X L. Differential detection and analysis of pathotypes ofpv.in hubei province. Hubei Agricultural Sciences, 2018, 57(24): 100-103. (in Chinese)
[31] 阎园园, 陈华, 姜波. 基于群智能算法分类模型的番茄病害识别. 江苏农业科学, 2020, 48(1): 219-224.
YAN Y Y, CHEN H, JIANG B. Tomato disease recognition based on swarm intelligence classification model. Jiangsu Agricultural Science, 2020, 48(1): 219-224. (in Chinese)
[32] 徐笑锋, 肖英杰, 章学来, 徐亚伟. 基于PCA-相对熵模型的海上中转引航平台选址研究. 安全与环境学报, 2021, 21(6): 2438-2443.
XU X F, XIAO Y J, ZHANG X L, XU Y W. Research on site selection of maritime transit pilot platform based on PCA-relative entropy model. Journal of Safety and Environment, 2021, 21(6): 2438-2443. (in Chinese)
[33] 吕开云, 鞠厦轶, 龚循强, 鲁铁定. 基于PCA和IGG权函数的人脸图像鲁棒线性回归分类方法. 电子测量技术, 2021, 44(21): 152-157.
LÜ K Y, JU X Y, GONG X Q, LU T D. Face recognition using robust linear regression classification based on PCA and IGG weight function. Electronic Measurement Technology, 2021, 44(21): 152-157. (in Chinese)
[34] 郭金玉, 刘玉超, 李元. 加权局部近邻标准化PCA的工业过程故障检测. 沈阳化工大学学报, 2021, 35(3): 265-274.
GUO J Y, LIU Y C, LI Y. Fault detection of industrial process based on weighed local neighborhood standardization PCA. Journal of Shenyang University of Chemical Technology, 2021, 35(3): 265-274. (in Chinese)
[35] REICH D, PRICE A L, PATTERSON N. Principal component analysis of genetic data. Nature Genetics, 2008, 40(5): 491-492.
[36] HUANG X, WEI X, SANG T, ZHAO Q, FENG Q, ZHAO Y, LI C, ZHU C, LU T, ZHANG Z,. Genome-wide association studies of 14 agronomic traits in rice landraces. Nature Genetics, 2010, 42(11): 961-967.
[37] ZHAO K, TUNG C W, EIZENGA G C, WRIGHT M H, ALI M L, PRICE A H, NORTON G J, ISLAM M R, REYNOLDS A, MEZEY J, MCCLUNG A M, BUSTAMANTE C D, MCCOUCH S R. Genome-wide association mapping reveals a rich genetic architecture of complex traits in. Nature Communications, 2011, 2: 467.
[38] JONNALAGADDA S, SRINIVASAN R. Principal components analysis based methodology to identify differentially expressed genes in time-course microarray data. BMC Bioinformatics, 2008, 9: 267.
[39] 周丽娜, 于海业, 张蕾, 任顺, 隋媛媛, 于连军. 基于叶绿素荧光光谱分析的稻瘟病害预测模型. 光谱学与光谱分析, 2014, 34(4): 1003-1006.
ZHOU L N, YU H Y, ZHANG L, REN S, SUI Y Y, YU L J. Rice blast prediction model based on analysis of chlorophyll fluorescence spectrum. Spectroscopy and Spectral Analysis, 2014, 34(4): 1003-1006. (in Chinese)
[40] WANG C Y, CHEN S, FENG A Q, SU J, WANG W J, FENG J Q, CHEN B, ZHANG M Y, YANG J Y, ZENG L X, ZHU X Y., a small orphan gene harboring promoter trap for AvrXa7, leads to the durable resistance topv. Rice, 2021, 14(1): 48.
[41] CHEN X F, LIU P C, MEI L, HE X L, CHEN L, LIU H, SHEN S R, JI Z D, ZHENG X X, ZHANG Y C, GAO Z Y, ZENG D L, QIAN Q, MA B J., a new executorgene that confers durable and broad-spectrum resistance to bacterial blight disease in rice. Plant Communications, 2021, 2(3): 100143.
[42] LUO D, HUGUET-TAPIA J C, RABORN R T, WHITE F F, YANG B. Theresistance gene guards the susceptibility geneof rice against exploitation by bacterial blight pathogen. Plant Communications, 2021, 2(3): 100164.
[43] JI C H, JI Z Y, LIU B, CHENG H, LIU H, LIU S Z, YANG B, CHEN G Y.allelicgenes activate rice blight resistance suppressed by interfering TAL effectors. Plant Communications, 2020, 1(4): 100087.
[44] ZHANG B M, ZHANG H T, LI F, OUYANG Y D, YUAN M, LI X H, XIAO J H, WANG S P. Multiple alleles encoding atypical NLRs with unique central tandem repeatsin rice confer resistance topv.. Plant Communications, 2020, 1(4): 100088.
[45] Pradhan S K, Barik S R, Sahoo A, Mohapatra S, Nayak D K, Mahender A, Meher J, Anandan A, Pandit E. Population structure, genetic diversity and molecular marker-trait association analysis for high temperature stress tolerance in rice. PLoS One, 2016, 11(8): e0160027.
[46] Baliyan N, Malik R, Rani R, Mehta K, Vashisth U, Dhillon S, Boora K S. Integrating marker-assisted background analysis with foreground selection for pyramiding bacterial blight resistance genes into Basmati rice. Comptes Rendus Biologies, 2018, 341(1): 1-8.
[47] Hsu Y C, Chiu C H, Yap R, Tseng Y C, Wu Y P. Pyramiding bacterial blight resistance genes in Tainung82 for broad-spectrum resistance using marker-assisted selection. International Journal of Molecular Sciences, 2020, 21(4): 1281.
[48] WANG Y, PRUITT R N, NÜRNBERGER T, WANG Y C. Evasion of plant immunity by microbial pathogens. Nature Reviews Microbiology, 2022, 20(8): 449-464.
Pathotype Analysis ofpv.in main Rice Producing Regions of China and Establishment of Differential Hosts of Near-isogenic Lines
FENG AiQing, WANG CongYing, ZHANG MeiYing, CHEN Bing, FENG JinQi, CHEN KaiLing, WANG WenJuan, Yang Jianyuan, Su Jing, Zeng LieXian, CHEN Shen, ZHU XiaoYuan
Plant Protection Research Institute, Guangdong Academy of Agricultural Sciences/Guangdong Provincial Key Laboratory of High Technology for Plant Protection, Guangzhou 510640
【Objective】The pathotypes ofpv.() in diverse rice regions in China were analyzed, and the differential hosts of near-isogenic lines (NILs) were established to provide a scientific basis for accurate field monitoring ofpopulation, application of resistant varieties and breeding of resistant varieties against rice bacterial blight.【Method】To explore virulence diversity and distribution of pathotypes of, the pathotypes of 954 single colony strains collected from Guangdong, Guangxi, Hainan, Zhejiang, Hunan, Liaoning and Yunnan provinces (Autonomous Region) from 2018 to 2021 were identified by artificial leaf-cutting inoculation on 21 differential hosts including Chinese differential hosts, IR24 and 15 bacterial blight NILs. Based on the resistant and susceptible interactions among the testedstrains, 15 NILs and IR24, the principal component analysis (PCA) was used to analyze the variable factors of which help abstraction of the candidate differentials. Based on the resistant and susceptible interactions between resistance genes and tested strains, the pyramiding effect of resistance genes was analyzed.【Result】The tested 954strains were divided into 11 pathotypes including SRRRR (I), SSRRR (Ⅱ), SSSRR (Ⅲ), SSSSR (Ⅳ), SSRRS (Ⅴ), SRSRR (Ⅵ), SSSSS (Ⅸ), SSSRS (new pathotype 1), SRSRS (new pathotype 2), SRSSS (new pathotype 3) and SSRSS (new pathotype 4) based on Chinese differential hosts. The percentage of each pathotype mentioned above was 11.53%, 4.82%, 7.34%, 6.18%, 7.23%, 1.05%, 59.96%, 1.57%, 0.10%, 0.10% and 0.10%, respectively. Pathotype Ⅸ, with broad pathogenicity and more virulence, had become the predominant race in the South China rice region and the Yangtze River rice region (Hunan, Zhejiang), and the predominant pathotypes in Yunnan (southwest China) and Liaoning (northeast China) were pathotype Ⅳ and pathotype I, respectively. The resistance and susceptibility of 15 rice NILs to 954strains were analyzed. The results showed that the 15 NILs could be divided into five types, highly susceptible lines with IRBB1, IRBB2, IRBB10, IRBB11, IRBB4, the moderately susceptible lines with IRBB3, IRBB203, IRBB14, moderately resistant lines with IRBB8, IRBB13, resistant lines with IRBB21, highly resistant lines with IRBB5, IRBB7, CBB23, GDBB23. Among the testedstrains, 42 strains could infect, 34 strains could infect, and 31 strains could infect. Factors were extracted from the interaction variable data matrix among 954 strains and 16 varieties (15 NILs and its recurrent parent IR24). with the total explained variable >85.0% as the boundary, eight principal components were extracted, and ten varieties (lines) mainly with NILs were constructed as differential hosts. Accord to their contribution to interaction variances, these differential hosts were IRBB10 (), IRBB4 (), GDBB23 (), IRBB5 (), IRBB7 (), IRBB21 (), IR24 (), IRBB13 (), IRBB3 (), Jingang30. With new differential hosts, 954 testedstrains could be divided into 55 pathotypes, the newly developed differential hosts showed good discrimination ability for monitoring the virulence ofin the tested rice regions. The results of gene pyramiding combined resistance analysis showed that the resistance frequency of different resistance genes pyramiding towas increased, and different resistance genes had diverse complementarity to the resistance of the test strains. 【Conclusion】Thestrains in the monitoring rice regions tended to be diversified, and the virulence differentiation was obvious. Pathotype Ⅸ of the virulent strain had become the prevailing races in some rice production regions, and the number of the strains compactible with broad-spectrum resistance genes,andshowed an increasing trend. Resistance gene polymerization can broaden the resistance spectrum of varieties to pathogen lines, which is an effective way to breed broad-spectrum resistant varieties. The establishment and application of NILs differential host could provide technical support for precise monitoring of the occurrence and early warning of epidemic of bacterial blight disease in the field.
pv.(); pathotype; near-isogenic line (NIL); differential host; resistance

10.3864/j.issn.0578-1752.2022.21.007
2022-05-10;
2022-06-03
国家水稻产业技术体系(CARS-01-35)、国家重点研发计划(2016YFD0300707)、广东省现代农业产业技术体系专项资金(2021KJ105)、广东省农业科学院学科团队建设项目(201613TD,202116TD)、广东省自然科学基金面上项目(2021A1515012497)、广东省农业科学院协同创新中心项目(XT202211)
冯爱卿,E-mail:fengaq@gdppri.com。通信作者朱小源,Tel:020-87597562;E-mail:zhuxy@gdppri.com
(责任编辑 岳梅)
猜你喜欢
今日农业(2022年4期)2022-06-01
中国稻米(2021年2期)2021-04-04
农民致富之友(2020年31期)2020-11-18
——稻(二)
种业导刊(2019年3期)2019-05-21
浙江农业学报(2017年1期)2017-05-17
上海农业学报(2017年3期)2017-04-10
上海农业学报(2017年3期)2017-04-10
种业导刊(2015年3期)2015-01-22
种业导刊(2014年3期)2014-01-23
现代农业科技(2009年17期)2009-03-08
