
蒲蓝利咽合剂的指纹图谱建立及含量测定Δ
2022-11-28 08:46:18王博于晓涛刘玉萍赫振玉王瑞漯河市中心医院药学部河南漯河462000河南省中药制剂与加工中医药重点实验室河南漯河462000河南省中药制剂现代化技术研发与临床应用工程研究中心河南漯河462000
中国药房 2022年22期
王博,于晓涛,刘玉萍,赫振玉,王瑞,3#(.漯河市中心医院药学部,河南 漯河 462000;2.河南省中药制剂与加工中医药重点实验室,河南 漯河 462000;3.河南省中药制剂现代化技术研发与临床应用工程研究中心,河南 漯河 462000)
蒲蓝利咽合剂由蒲公英、黄芩、木蝴蝶、忍冬藤等6味药材组成,由《千家妙方》记载的“复方蒲公英汤”[1]加减化裁而来,能透泄里热壅盛之邪,具有清热解毒、凉血利咽之效,主要用于治疗上呼吸道感染引起的发热、咽喉肿痛等症。方中蒲公英为君药,清热解毒、消肿散结[2];黄芩为臣药,善清上焦之火,可治火毒炽盛的疮痈肿毒、咽喉肿痛[3];木蝴蝶、忍冬藤等为佐使药,疏风通络、清肺利咽[4―5];诸药共奏清热解毒、抗炎消肿之效。蒲蓝利咽合剂主要含有酚酸类、黄酮类、萜类、挥发油等成分,其中绿原酸、咖啡酸、菊苣酸为君药蒲公英中酚酸类成分,具有抗菌、抗病毒、抗炎等作用[6―8];黄芩苷、千层纸素A-7-O-β-D-葡萄糖醛酸、汉黄芩苷为臣药黄芩中黄酮类成分,具有抗病毒、神经保护等作用[9];马钱苷为佐药忍冬藤中环烯醚萜类成分,具有抗氧化、抗菌及增强免疫功能等作用[10];木蝴蝶苷B为使药木蝴蝶的特征性成分,具有抗菌、抗炎等作用[11]。目前,对蒲蓝利咽合剂的质量控制仅限于薄层鉴别及相关检查项,尚缺少定量控制的方法。
由于中药成分复杂,对一种或几种成分的定量研究不能全面反映中药制剂的整体质量。中药指纹图谱所含化学成分信息量大、特征性强,能对中药质量进行整体描述和评价[12],结合化学模式识别能对指纹图谱的整体性和模糊性进行更加科学合理的评价[13]。基于此,本研究建立了蒲蓝利咽合剂的高效液相色谱(HPLC)指纹图谱,并进行化学模式识别分析;同时采用相同的HPLC法测定该制剂中绿原酸、咖啡酸、马钱苷、菊苣酸、木蝴蝶苷B、黄芩苷、千层纸素A-7-O-β-D-葡萄糖醛酸、汉黄芩苷共8种活性成分的含量,旨在为蒲蓝利咽合剂的质量评价提供参考。
1 材料
1.1 主要仪器
本研究所用主要仪器有LC-20A型HPLC仪、AUW-120D型十万分之一天平(日本Shimadzu公司),Milli-Q型纯水仪(美国Millipore公司),KQ-300E型超声波清洗器(昆山市超声仪器有限公司)。
1.2 主要药品与试剂
对照品绿原酸(批号110753-202119,纯度96.8%)、咖啡酸(批号110885-201703,纯度99.7%)、黄芩苷(批号110715-202122,纯度95.4%)、汉黄芩苷(批号112002-201702,纯度98.5%)均购自中国食品药品检定研究院;对照品千层纸素A-7-O-β-D-葡萄糖醛酸(批号DSTDQ-004001,纯度98.49%)、马钱苷(批号DSTDM003801,纯度99.31%)、菊苣酸(批号DSTDJ003901,纯度98.23%)、木蝴蝶苷B(批号DSTDM002901,纯度98.39%)均购自成都德思特生物科技有限公司;乙腈、甲酸均为色谱纯,水为纯化水。
11批蒲蓝利咽合剂[规格为每毫升含生药0.52 g(100 mL/瓶),编号 S1~S11,批号分别为 20210621、20220214、20220216、20220218、20220221、20220223、20220225、20220228、20220302、20220304、20220307]均为漯河市中心医院自制。
黄芩饮片(批号20201201)、忍冬藤饮片(批号20191101)、木蝴蝶饮片(批号20201001)均购自安徽桐花堂中药饮片科技有限公司,蒲公英饮片(批号180601)购自亳州市万草堂中药饮片有限公司,均经漯河市中心医院制剂室于晓涛副主任中药师鉴定为真品。
2 方法与结果
2.1 色谱条件
以 Shimadzu Shim-pack GIST C18(150 mm×4.6 mm,5 μm)为色谱柱,以0.1%甲酸溶液(A)-乙腈(B)为流动相进行梯度洗脱(0~7 min,7%B;7~15 min,7%B→8%B;15~27 min,8%B→10%B;27~32 min,10%B→15%B;32~45 min,15%B;45~55 min,15%B→19%B;55~60 min,19%B→22%B;60~80 min,22%B→25%B;80~85 min,25%B→40%B;85~100 min,40%B→50%B;100~105 min,50%B→90%B);柱温为25 ℃;流速为1.0 mL/min;进样量为5 μL;指纹图谱检测波长为254 nm;含量测定检测波长为237 nm(马钱苷)、280 nm(木蝴蝶苷B、黄芩苷、千层纸素A-7-O-β-D-葡萄糖醛酸、汉黄芩苷)、327 nm(绿原酸、咖啡酸、菊苣酸)。
2.2 溶液的制备
2.2.1 混合对照品溶液 取绿原酸、咖啡酸、马钱苷、菊苣酸、木蝴蝶苷B、黄芩苷、千层纸素A-7-O-β-D-葡萄糖醛酸、汉黄芩苷对照品适量,置于10 mL容量瓶中,加50%甲醇制成上述各成分质量浓度分别为68.400、30.000、250.625、237.600、105.600、2 204.800、289.300、408.000 μg/mL的混合对照品溶液。
2.2.2 供试品溶液 取蒲蓝利咽合剂1 mL,加水稀释至10 mL,以14 000 r/min离心10 min,取上清液,即得供试品溶液。
2.2.3 阴性样品溶液 按蒲蓝利咽合剂处方工艺分别制备缺黄芩或蒲公英或忍冬藤或木蝴蝶以及同时缺蒲公英和忍冬藤(因两者均含有咖啡酸)、黄芩和木蝴蝶(因两者均含有黄芩苷)的阴性样品,按“2.2.2”项下方法制备各阴性样品溶液。
2.3 蒲蓝利咽合剂HPLC指纹图谱的建立
2.3.1 精密度试验 精密量取编号为S11的供试品溶液,按“2.1”项下色谱条件连续进样6次,记录峰面积。以黄芩苷为参照峰,结果显示,各共有峰相对保留时间和相对峰面积的RSD分别为0.010%~0.234%、0.083%~2.935%(n=6),表明方法精密度良好。
2.3.2 重复性试验 精密量取编号为S11的样品,共6份,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样,记录峰面积。以黄芩苷为参照峰,结果显示,各共有峰相对保留时间和相对峰面积的RSD分别为0.025%~0.481%、0.140%~2.777%(n=6),表明方法重复性良好。
2.3.3 稳定性试验 精密量取编号为S11的供试品溶液,分别于室温下放置0、2、4、8、12、24 h时按“2.1”项下色谱条件进样,记录峰面积。以黄芩苷为参照峰,结果显示,各共有峰相对保留时间和相对峰面积的RSD分别为0.034%~0.630%、0.272%~2.865%(n=6),表明供试品溶液于室温下放置24 h内稳定性良好。
2.3.4 指纹图谱的生成和相似度评价 取11批样品,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样,记录色谱峰。将所得的色谱图采用《中药色谱指纹图谱相似度评价系统(2012版)》软件进行分析,以峰面积适中的S11样品为对照图谱,选择中位数计算,采用多点校正生成HPLC叠加指纹图谱和对照指纹图谱(R)。结果显示,11批样品共有20个共有峰,详见图1。11批样品的相似度分别为0.988、0.990、0.991、0.985、0.991、0.989、0.973、0.981、0.989、0.994、0.993。
2.3.5 共有峰指认 经过与相应对照品图谱(图2)比对,共指认出8个色谱峰,分别为绿原酸(5号峰)、咖啡酸(7号峰)、马钱苷(8号峰)、菊苣酸(9号峰)、木蝴蝶苷B(13号峰)、黄芩苷(14号峰,由于其保留时间居中,将其作为参照峰)、千层纸素A-7-O-β-D-葡萄糖醛酸(17号峰)、汉黄芩苷(18号峰)。
2.4 HPLC指纹图谱的化学模式识别分析
2.4.1 聚类分析 将11批样品的20个共有峰相对峰面积导入SPSS 20.0软件,采用组间联结法,以平方欧氏距离为区间进行聚类分析。结果显示,当平方欧氏距离为10时,11批样品分为2大类,其中S3、S10~S11为一类,其余为一类。结果见图3。
2.4.2 主成分分析 以11批样品的20个共有峰相对峰面积为变量,运用SPSS 20.0 软件进行主成分分析(表1)。结果显示,前4个主成分的累计方差贡献率为95.546%,表明这4个主成分能够反映指纹图谱共有峰的主要信息。采用SIMCA14.1软件进行无监督模式的主成分建模分析(图4)。结果显示,11批样品分为2类,S3、S10~S11为一类,其余为一类,与聚类分析结果一致。

表1 4个主成分因子的特征值和方差贡献率
2.4.3 正交偏最小二乘法-判别分析 以11批样品的20个共有峰相对峰面积为变量,运用SIMCA14.1软件进行正交偏最小二乘法-判别分析(图5)。结果显示,所得模型参数RX2=0.891,RY2=0.963,Q2=0.899>0.5,表明模型稳定,预测能力良好。
以变量重要性投影(variable importance in projec‐tion,VIP)值大于1[14]为指标筛选对样品分类贡献较大的成分(图6)。结果显示,15、14(黄芩苷)、10、18(汉黄芩苷)、11、17(千层纸素A-7-O-β-D-葡萄糖醛酸)、4、20、16、12、7(咖啡酸)、19号峰的VIP值大于1,表明这12个共有峰对应的成分可能是影响蒲蓝利咽合剂质量的标志物。
2.5 蒲蓝利咽合剂中绿原酸等8种成分的含量测定
采用相同的HPLC法测定蒲蓝利咽合剂中绿原酸等8种活性成分的含量。
2.5.1 专属性试验 取“2.2”项下混合对照品溶液、供试品溶液(编号S11)、各阴性样品溶液,按“2.1”项下色谱条件进样,记录色谱图(图2,阴性样品图略)。结果显示,各成分分离度良好,阴性样品无干扰。
2.5.2 线性关系考察 精密量取“2.2.1”项下混合对照品溶液0.1、0.2、0.5、1.0、2.0、3.0 mL,分别置于5 mL容量瓶中,加50%甲醇稀释,得系列浓度工作溶液。取系列浓度工作溶液及混合对照品溶液,按“2.1”项下色谱条件进样,记录色谱峰。以各待测成分质量浓度为横坐标(x)、峰面积为纵坐标(y)进行线性回归。结果见表2。

表2 绿原酸等8种成分的回归方程与线性范围
2.5.3 精密度试验 精密量取“2.2.1”项下混合对照品溶液1 mL,置于5 mL容量瓶中,加50%甲醇定容至刻度,按“2.1”项下色谱条件连续进样6次,记录峰面积。结果显示,绿原酸、咖啡酸、马钱苷、菊苣酸、木蝴蝶苷B、黄芩苷、千层纸素A-7-O-β-D-葡萄糖醛酸、汉黄芩苷峰面积的RSD分别为0.60%、0.67%、0.65%、0.60%、0.47%、0.54%、0.52%、0.54%(n=6),表明仪器精密度良好。
2.5.4 重复性试验 精密吸取样品(编号S11),共6份,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样,记录峰面积并按标准曲线法计算样品含量。结果显示,绿原酸、咖啡酸、马钱苷、菊苣酸、木蝴蝶苷B、黄芩苷、千层纸素A-7-O-β-D-葡萄糖醛酸、汉黄芩苷含量的RSD分别为0.79%、0.27%、0.22%、0.12%、0.68%、0.49%、0.08%、0.12%(n=6),表明方法重复性良好。
2.5.5 稳定性试验 取“2.2.2”项下供试品溶液(编号S11),分别于室温下放置0、2、4、8、12、24 h时按“2.1”项下色谱条件进样,记录峰面积。结果显示,绿原酸、咖啡酸、马钱苷、菊苣酸、木蝴蝶苷B、黄芩苷、千层纸素A-7-O-β-D-葡萄糖醛酸、汉黄芩苷峰面积的RSD分别为1.02%、0.69%、0.54%、0.35%、0.16%、0.32%、0.30%、0.26%(n=6),表明供试品溶液于室温下放置24 h内稳定。
2.5.6 加样回收率试验 精密量取样品(编号S11)0.5 mL,共6份,按1∶1加入混合对照品溶液,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样,记录峰面积并计算加样回收率。结果显示,绿原酸、咖啡酸、马钱苷、菊苣酸、木蝴蝶苷B、黄芩苷、千层纸素A-7-O-β-D-葡萄糖醛酸、汉黄芩苷的平均加样回收率分别为100.69%、98.50%、101.86%、99.97%、104.34%、100.38%、101.84%、99.74%,RSD 分别为 1.99%、2.64%、1.42%、1.54%、2.89%、2.73%、1.53%、1.25%(n=6),表明方法准确度良好。
2.5.7 样品含量测定 精密量取11批样品,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样,记录峰面积并按标准曲线法计算样品含量,每批样品平行测定3次,取平均值。结果见表3。
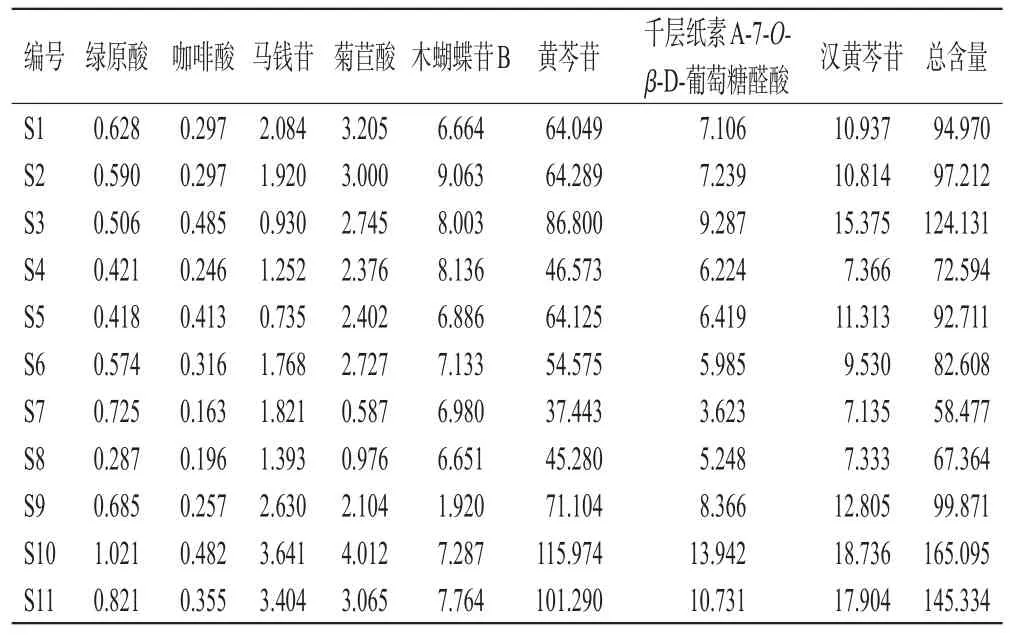
表3 绿原酸等8种成分的含量测定结果(n=3,mg/g)
3 讨论
本课题组前期分别考察了以甲醇-甲酸溶液、乙腈-甲酸溶液、乙腈-乙酸溶液为流动相时各成分的分离效果,结果表明,以乙腈-甲酸溶液为流动相时各成分的分离效果较好,故选择其为流动相。同时还考察了不同波长下(230、254、280、330 nm)各色谱峰的吸收度,结果显示,在254 nm波长下所得色谱峰较多且分离度较好,能够较为全面地反映样品的化学成分信息,故选择254 nm为指纹图谱的检测波长;此外还对绿原酸等8种成分进行全波长扫描,结果发现,绿原酸、咖啡酸、菊苣酸的最大吸收波长为327 nm,马钱苷为237 nm,木蝴蝶苷B、黄芩苷、千层纸素A-7-O-β-D-葡萄糖醛酸、汉黄芩苷均为280 nm,因此选择237、280、327 nm为含量测定的检测波长。
本研究建立了11批蒲蓝利咽合剂的HPLC指纹图谱,确定了20个共有峰,与对照指纹图谱的相似度均大于0.97,说明各批次样品的质量均一稳定、差异较小。聚类分析、主成分分析的结果均显示,S3、S10~S11为一类,其余为一类。正交偏最小二乘法-判别分析结果显示,15、14(黄芩苷)、10、18(汉黄芩苷)、11、17(千层纸素A-7-O-β-D-葡萄糖醛酸)、4、20、16、12、7(咖啡酸)、19号峰为影响样品质量的标志物。考虑到复方制剂具有多成分、多靶点协同作用的特点,故选择样品中发挥抗菌消炎作用的主要药效成分绿原酸、菊苣酸、马钱苷和木蝴蝶苷B[6,8,10―11],与 VIP 值>1 的黄芩苷、千层纸素 A-7-O-β-D-葡萄糖醛酸、汉黄芩苷、咖啡酸共8种成分作为蒲蓝利咽合剂质量控制的指标成分。
含量测定结果显示,11批样品中绿原酸、咖啡酸、马钱苷、菊苣酸、木蝴蝶苷B、黄芩苷、千层纸素A-7-O-β-D-葡萄糖醛酸、汉黄芩苷的含量分别为0.287~1.021、0.163~0.485、0.735~3.641、0.587~4.012、1.920~9.063、37.443~115.974、3.623~13.942、7.135~18.736 mg/g,表明不同批次样品含量存在差异,其中S3、S10~S11样品中8种成分的总含量高于其他批次。其原因可能与蒲蓝利咽合剂原药材的产地、制剂提取浓缩工艺等多种因素有关[15―16]。
综上所述,建立的蒲蓝利咽合剂HPLC指纹图谱和绿原酸等8种成分的含量测定方法,可用于蒲蓝利咽合剂的质量评价;黄芩苷、汉黄芩苷、千层纸素A-7-O-β-D-葡萄糖醛酸等12种成分可能是影响蒲蓝利咽合剂质量的标志物。
猜你喜欢
保健与生活(2023年22期)2023-12-26 07:29:35
家庭医药(2021年3期)2021-04-12 15:09:05
河北果树(2020年2期)2020-05-25 06:58:58
天然产物研究与开发(2019年1期)2019-03-01 05:41:08
中成药(2018年11期)2018-11-24 02:56:46
中成药(2018年5期)2018-06-06 03:11:54
中成药(2017年7期)2017-11-22 07:33:11
中成药(2017年8期)2017-11-22 03:19:32
中成药(2017年10期)2017-11-16 00:50:42
中国中医药现代远程教育(2014年19期)2014-03-01 04:30:46
