
基于外切酶驱动策略构建荧光减弱式T4 PNK免标记传感器
2022-11-19 07:02刘业玲刘炳鑫黄圣荣周冰倩薛庆旺
聊城大学学报(自然科学版) 2022年2期
刘业玲,袁 珲,刘炳鑫,黄圣荣,周冰倩,李 霞,薛庆旺
(聊城大学 化学化工学院,山东 聊城 252059)
0 引言
T4多核苷酸激酶(T4 PNK)是PNK家族的主要成员,属于DNA修复酶,其功能是将三磷酸腺苷(ATP)的γ位转移到核酸的5′-OH端,以催化核酸的磷酸化过程[1-3]。T4 PNK在DNA损伤修复和维持基因组稳定性中起关键作用,许多研究证明:T4 PNK活性的异常与人类多种恶性疾病密切有关[4-6]。因此T4 PNK活性的灵敏检测对临床诊断和疾病治疗至关重要。
目前,对T4 PNK活性分析检测传统方式有放射性同位素32P标记[7],聚丙烯酰胺凝胶电泳[8]等方式,但这些检测方法存在操作复杂,灵敏度低等不足的缺点。为此,具有更高灵敏度的电化学[9]、比色[10]、荧光[11]检测方法陆续被开发。其中,通过荧光发射强度为信号的荧光分析方法以其简单、快速、信号转导模式灵活而被科研工作者所青睐[12]。核酸探针基于它的碱基互补配对原则以及良好的生物相容性备受关注。在常用的荧光检测体系中,大多数报道的信号输出探针以荧光标记型核酸探针为主[11,13],但将荧光基团共价键合到DNA末端的操作过程复杂,需要专业的技术人员合成并对其进行复杂的HPLC纯化流程,实验成本较高,因此限制了标记型探针的广泛应用。为此,各种成本低廉的免标记荧光探针被逐渐开发。通过荧光染料分子如:硫黄素T(ThT)[14]、锌卟啉(ZnPPIX)、SYBR Green Ι(SG Ι)[15],其中,鉴于游离态SYBR Green Ι(SG Ι)荧光染料分子几乎不产生荧光,并且对DNA序列没有要求,当与双链DNA特异性结合后激活出强烈稳定免标记荧光信号,因此DNA-SG免标记信号输出探针逐渐引起研究人员的广泛关注。虽然DNA-SG Ι复合物为低背景、免标记荧光分析提供了新契机,但所涉及的基底探针以双链DNA或发夹DNA为主,双链部分处于未封闭状态,即使在靶标不存在情况,仍能产生较高的背景信号。因此构建一种低背景的免标记荧光方法,对于提高T4 PNK活性测定灵敏度具有重要意义。
在这里,我们提出了基于λ核酸外切酶驱动荧光减弱式检测T4 PNK活性的策略。首先,我们采用了两种类型的DNA探针sg和msg。二者可以杂交形成无任何标记的双链DNA(dsDNA)。在没有T4 PNK的存在下,加入λ核酸外切酶(λexo)和核酸外切酶Ι(Exo Ι)之后不会对底物dsDNA产生影响,加入与dsDNA特定结合的荧光染料SG Ι后产生较强荧光;然而在靶标T4 PNK存在的情况下,dsDNA的5′端由羟基变为磷酸基团,而λexo催化dsDNA分子从5′-P末端进行逐步的水解释放出5′-单核苷酸。但不能降解5′-OH末端。之后加入Exo Ι 持续切割另一条单链sg,体系中的dsDNA减少,导致与荧光染料SG Ι键合位点减少,其荧光强度被大大减少。因此,可以利用荧光减弱的程度来实现对T4 PNK的免标记荧光检测。
1 实验部分
1.1 实验药品
T4多核苷酸激酶、λ核酸外切酶、核酸外切酶Ι、三磷酸腺苷、RNase-free water、牛血清白蛋白(BSA)、SYBR Green Ι(SG Ι)、上样缓冲液(Loading Buffer)、凝胶核酸染料Super Gelblue均购买来自New England Biolabs公司(北京)。脱氧核苷酸溶液混合物(dNTPs)、三磷酸腺苷(ATP)和所有HPLC纯化的寡核苷酸序列均由生工生物工程股份有限公司(上海,中国)获得;所涉及的寡核苷酸序列如下:具有5′-OH-TAA TAC GAC TCA CTA TA GGG TAA TTT CTA CTA AGT GTA GAT GAG AAG TCA TCT AAT AAG GCG-OH -3′的序列的DNA探针(表示为DNA-sg);具有5′-OH-CGC CTT ATT AGA TGA CTT CTC ATC TAC ACT TAG TAG AAA TTA CCC TAT AGT GAG TCG TAT TA-OH-3′序列的引物探针(表示为DNA-msg); 其他化学试剂(分析纯)均购自标准试剂提供商。实验中所用水均为高压灭菌过的高纯水(≥18.2 MΩ)
1.2 仪器
Milli-Q 超纯水处理系统(美国 Millipore);立式压力蒸汽灭菌器(LDZX-50KBS,上海申安医疗器械厂);真空干燥箱(DZ-ZA型,天津市泰斯特仪器有限公司);干式恒温器(杭州奥盛仪器有限公司) ;凝胶成像仪(Bio-Rad,Gel Doc XR+,美国)、电泳仪(DYY-6C型,北京六一仪器厂)。所有荧光强度的测量均使用F-7000荧光分光光度计(日本,日立),激发波长设定为490 nm,发射波长设定为510~650 nm,激发狭缝为5 nm,发射狭缝为10 nm,PMT检测电压为650 V。
1.3 实验步骤
将两种类型的DNA探针sg和msg于37 ℃下在1X T4 PNK Buffer中孵育2 h形成dsDNA(浓度为2.1 μmol/L)。取5 μL dsDNA之后加入3 μL 10 mmol/L ATP和一定量的T4 PNK于37 ℃下在1X T4 PNK Buffer中孵育2 h。自然冷却至室温后,分别加入1 μL 5 Uλexo,1.5 μLλexo Buffer,0.5 μL 20 U Exo Ι和1.5 μL Exo Ι Buffer,在37 ℃下孵育2 h,80 ℃下反应20 min对上述酶灭活。自然冷却至室温后加入10 μL SG Ι在37 ℃下避光反应30 min。自然冷却至室温,加水稀释到100 μL进行荧光光谱表征。最后,用12%非变性聚丙烯酰胺(PAGE)凝胶电泳分析产物,其条件在1X TBE缓冲液(9 mmol/L Tris-HCl,pH 7.90,9 mmol/L H3BO3,0.2 mmol/L EDTA),20 ℃、120 V恒定电压下进行。获得的凝胶用Super Gelblue染色并使用凝胶成像仪拍摄。
1.4 实际样品检测
为评估所提出的方法在实际样品中的应用效果,对细胞裂解液样品中的T4 PNK进行了检测分析。所用细胞裂解液为Hela细胞的裂解液。在分析之前,将细胞裂解液样品稀释1000倍,之后,往细胞裂解液样本中分别加入一定量的T4 PNK进行分析,分析步骤与上述过程相同。
2 结果与讨论
2.1 实验基本原理
本文所提出的外切酶驱动的荧光减弱式T4 PNK传感器原理如图1所示。在提出的检测策略中,以两种类型的DNA探针(sg和msg)杂交形成无任何标记的双链DNA(dsDNA)作为基底探针单元。由于SG Ι在游离态或与单链DNA(ssDNA)键合时荧光信号非常弱,但当它嵌入到双链DNA时荧光信号显著增强。当T4 PNK不存在时,msg探针的5′-OH末端未被磷酸化保持完整的5′-OH末端。随后加入的λexo和Exo Ι无法降解dsDNA。因此,当加入与dsDNA结合的荧光染料SG Ι后产生较强荧光。相反,在靶标T4 PNK存在的情况下,msg探针的5′末端被磷酸化,并产生具有λexo特定的识别位点-磷酸基团。因此,λexo催化dsDNA分子从5′-P末端进行逐步的水解释放出5′-单核苷酸,后续加入的Exo Ι继续降解单链DNA-sg,体系中的dsDNA减少导致与荧光染料SG Ι键合位点减少,其荧光强度被大大减弱。因此,利用荧光减弱的程度可实现对T4 PNK的免标记荧光检测。
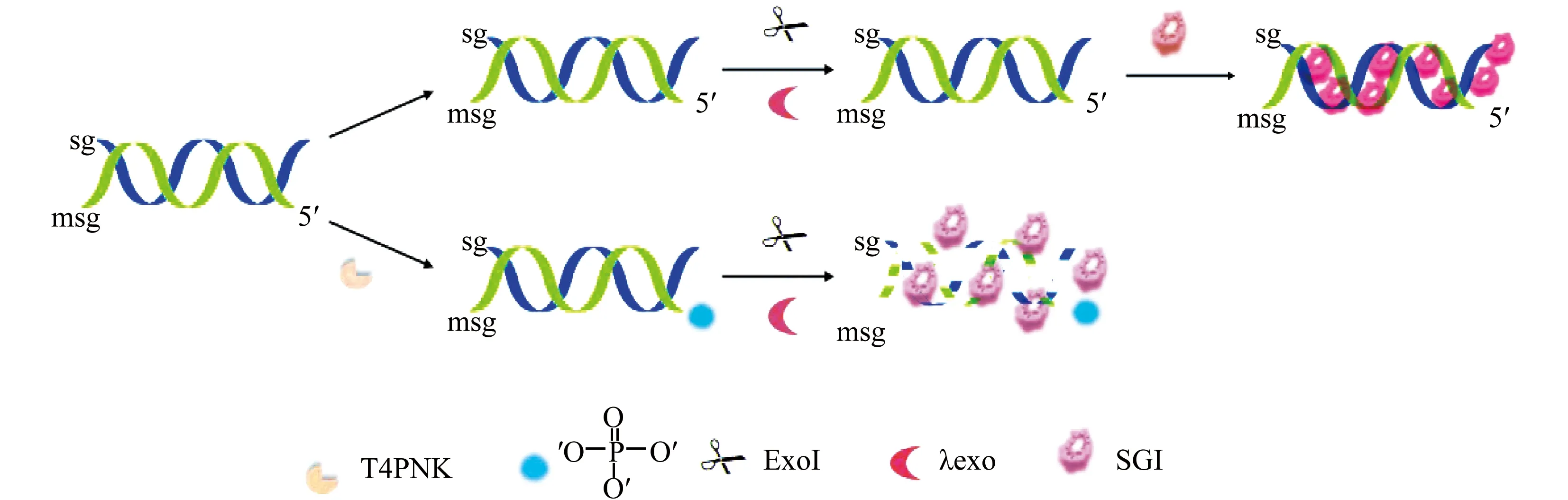
图1 基于外切酶驱动的荧光减弱式T4 PNK传感器原理图
2.2 可行性验证
为验证本工作所提出的T4 PNK检测策略的可行性,首先进行了荧光光谱分析。如图2所示,在T4 PNK不存在的情况下,系统显示出非常强的荧光信号(图2,曲线a)。这表明:λexo对5′-OH末端基团不起作用,无法产生ssDNA,Exo Ι不消化dsDNA,dsDNA仍然保持完整,因此加入SG Ι后嵌入到dsDNA时荧光信号显著增强。在T4 PNK存在的对照体系中,系统显示出微弱的荧光信号(图2,曲线b)。这是因为:5′-OH末端被磷酸化,并产生具有λexo特定的识别位点-磷酸基团。因此,λexo催化双链DNA分子从5′-P末端进行逐步的水解释放出5′-单核苷酸,生成单链DNA-sg,加入的Exo Ι继续降解单链DNA-sg,体系中的dsDNA减少导致与荧光染料SG Ι键合位点减少,其荧光强度被大大减弱。综合上述结果表明,通过荧光光谱表征证明了本检测方案具有良好的可行性。

图2 不同条件下的荧光光谱验证所构筑的T4 PNK传感器的可行性(曲线a)在T4 PNK(10 U·mL-1)存在下;(曲线b)在没有T4 PNK存在下;λexo,50 U·mL-1;Exo Ι,200 U·mL-1;SG I(10X),10 μL
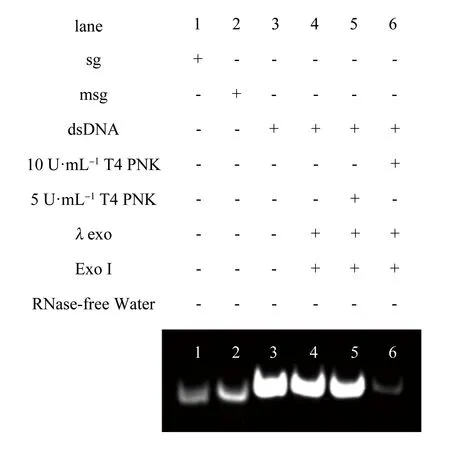
图3 聚丙烯酰胺凝胶电泳表征图
因此,为了进一步验证所提出的基于外切酶驱动的荧光减弱式传感器的可行性,进行了非变性PAGE实验。泳道1和泳道2分别对应sg与msg,因此只有单一条带作为参与反应的初始链存在,二者可以通过碱基互补配对原则进行杂交生成dsDNA(泳道3),因sg与msg的杂交产物,分子量大于sg与msg单链,因此位置高于泳道1和泳道2,泳道3表明sg与msg成功杂交形成dsDNA分子量增加,因此出现一条位置靠上的新的条带。在目标T4 PNK存在时,dsDNA的5′-OH末端被磷酸化,产生具有λexo特定的识别位点-磷酸基团,在λexo的作用下被识别降解,同时产生可被Exo Ι识别降解的DNA链,最后在Exo Ι作用下降解DNA单链,导致体系中的dsDNA被降解,条带明显变浅(泳道5、6);而在没有T4 PNK存在的情况下,dsDNA的5′-OH末端保持不变,λexo与Exo Ι无法进行作用,dsDNA保持不变(泳道4)。非变性PAGE结果进一步证实了所提出的基于外切酶驱动的荧光减弱式传感器是可行性。
2.3 条件优化
磷酸化反应时间和外切酶对磷酸化DNA的酶切效果会影响传感系统的荧光强度。为探索该方法的最佳反应条件,我们利用单因素变量法对λexo、Exo Ι和磷酸化反应时间分别进行了优化。如图4(a)所示,荧光强度的变化随着λexo含量的增加,荧光强度不断减弱。当达到4 U时,荧光强度降至最低达到平台。在平台期,λexo的最佳用量记录为4 U。如图4(c)所示,随着Exo Ι的不断增加,荧光强度出现一个低谷,此时的Exo Ι用量为15 U。因此,将Exo Ι的最佳反应用量选择15 U。T4 PNK可以使dsDNA的5′端由羟基化变为磷酸化,T4 PNK与dsDNA作用时间的长短决定体系中dsDNA磷酸化的程度。随着T4 PNK与dsDNA的作用时间的延长,荧光信号发生变化。当T4 PNK与dsDNA的作用时间为40 min时,荧光减弱的程度最多。最终,选择40 min作为最佳反应时间(图4(b))。因此根据图4的实验结果,最终选择λexo的用量、Exo Ι的用量和磷酸化反应时间分别为 4 U、15 U和40 min 作为反应体系的最佳反应条件。

图4 (a)λexo用量变化对荧光强度的影响;(b)磷酸化反应时间对荧光强度的影响;(c)Exo Ι用量变化对荧光强度的影响
2.4 传感器的线性范围分析
在上述最佳反应条件下,我们评估了本工作提出的基于外切酶驱动的荧光减弱式传感器对T4PNK灵敏检测的灵敏度。如图5(a)和(b)所示,发现荧光强度与T4PNK的浓度在0.01~5.00U·mL-1的范围内线性良好,T4PNK浓度的线性函数为ΔF=3265.29×logC+5691.71(C为T4PNK的浓度;R2=0.99945),检测下限为0.0173U·mL-1,所得的检测限优于和跟之前文献报道的结果相当。
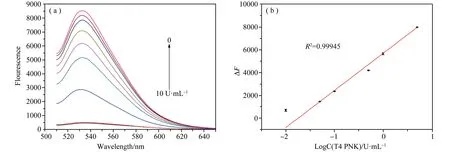
图5 (a) 不同浓度的T4 PNK所对应的荧光强度-波长曲线,T4 PNK浓度由0~10 U·mL-1;(b) 荧光强度与T4 PNK浓度的线性范围关系图

表1 不同方法检测T4 PNK酶活性的比较
2.5 传感选择性考察
为了评估T4 PNK传感器的选择性,在相同的实验条件下,我们研究了灭活后的T4 PNK,牛血清白蛋白(BSA)、Klenow片段聚合酶(KFP)等几种非特异性蛋白对体系的干扰。实验结果如图6所示,在T4 PNK存在的情况下,荧光强度发生较明显的降低。而在灭活后的T4 PNK存在时,荧光强度没有明显的下降,因为体系中dsDNA没有磷酸化反应的发生,无法触发之后的切割反应,导致体系中存在较多的dsDNA与SG Ι相互作用,发出强烈的荧光信号。同样,在BSA和KFP存在时,也未检测到明显的荧光信号降低。这一结果表明该方法对T4 PNK检测具有良好的选择性。
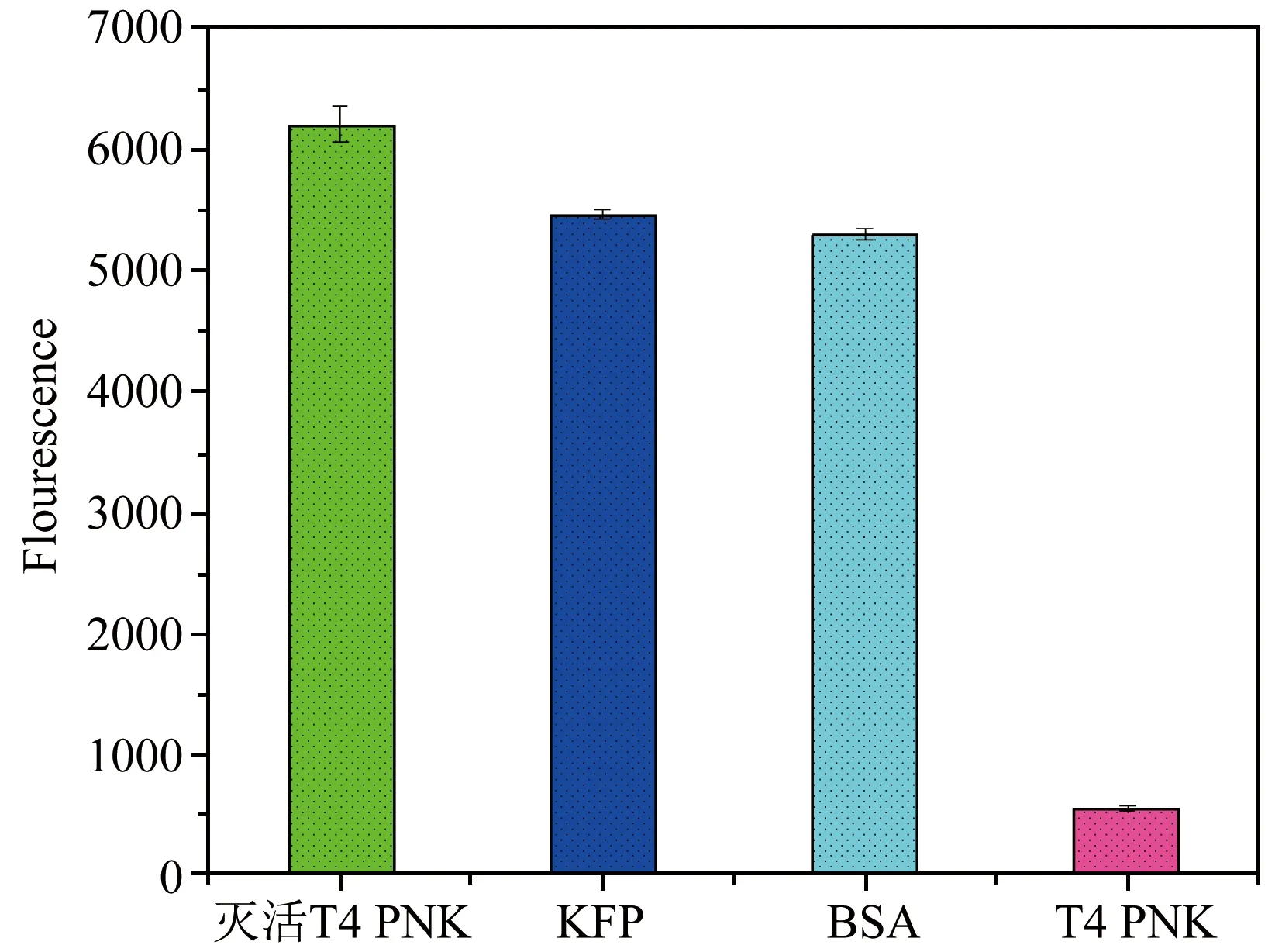
图6 基于所构筑的T4 PNK传感器选择性考察(灭活T4 PNK、KFP、BSA、T4 PNK浓度均为10 U·mL-1)
2.6 实际样品分析
为了证明该传感方法在实际样品测定中的应用前景,对细胞裂解液样品中的T4 PNK进行了研究。结果表明回收率为104.7%,并且相应的RSD值小于7.5%。因此,该方法在复杂的生物样品中可以很好地工作,在实际样品分析中具有很大的应用潜力。
3 结论
开发了一种新的基于外切酶驱动的荧光减弱式传感器对T4 PNK的灵敏检测。在我们的策略中,利用λexo、Exo Ι的切割反应,成功地建立了一种无标记、灵敏的荧光测定T4 PNK活性的方法。该方案显示出对T4 PNK检测的良好分析性能,具有较高的灵敏度、特异性和可重复性。在最佳条件下,测得线性范围在0.01~5.00 U·mL-1。并且与其他基于荧光的策略相比,所提出的方法具有以下的优点:它不需要使用荧光染料或用荧光团、淬灭剂对其标记,使得DNA探针的设计具有成本效益且简单;并且该种方法具有普适性,对任何序列的DNA都适用。此策略为探测T4 PNK活动提供了一种非常简便的方法,有望应用于生物医学研究。
猜你喜欢
中风与神经疾病杂志(2022年9期)2022-10-19
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
当代化工(2020年2期)2020-03-18
小资CHIC!ELEGANCE(2018年28期)2018-09-14
分析化学(2017年12期)2017-12-25
分析化学(2016年12期)2017-02-04
小资CHIC!ELEGANCE(2016年15期)2016-07-26
卷宗(2014年7期)2014-08-27
卷宗(2014年1期)2014-03-20
