
进行性家族性肝内胆汁淤积3型1例报告
2022-11-16 08:01张恬莹刘卫辉李贞茂李良平
临床肝胆病杂志 2022年11期
张恬莹,周 超,刘卫辉,李贞茂,李良平,雷 蕾
1 成都市金牛区人民医院 消化内分泌科,成都 610036;2 四川省医学科学院·四川省人民医院 消化内科,成都 610072
进行性家族性肝内胆汁淤积症(progressive familial intrahepatic cholestasis,PFIC)是常染色体隐性遗传性胆汁淤积性肝病,主要由基因突变后所致胆汁分泌或排泄障碍,随着病情的发展,最后可进展为肝纤维化、肝硬化、肝衰竭[1]。根据基因突变不同,PFIC目前分为6种亚型,分别由ATP8B1、ABCB11、ABCB4、TJP2、NR1H4和MYO5B基因突变导致[2],现将四川省人民医院消化为科收治的1例表现为肝内胆汁淤积的患者,经基因检测、肝组织活检、影像学等证实为PFIC3型患者的临床资料报道如下。
1 病例资料
患者男性,41岁,因“反复上腹痛,黄疸1个月余”于2021年2月18日收入本科,患者于入院前1个多月无明显诱因出现上腹部疼痛,呈持续性胀痛,无恶心、呕吐、腹泻、便血、呕血等体征,入院前外院腹部MRI提示胆总管结石,患者先于本院肝胆外科住院,2021年1月15日肝功能:AST 155 U/L、ALT 351 U/L、ALP 198 U/L、GGT 255 U/L、TBil 147.2 μmol/L、DBil 100.3 μmol/L、IBil 46.9 μmol/L,予以复方二氯酸醋酸二异丙胺注射液保肝、头孢哌酮钠他唑巴坦钠抗感染、补液等治疗,患者于2021年1月19日行ERCP+EST,术后腹痛缓解出院,出院后继续以熊去氧胆酸(UDCA)退黄治疗。2021年1月28日复查肝功能:AST 58 U/L、ALT 158 U/L、ALP 136 U/L、GGT 96 U/L、TBil 145 μmol/L、DBil 112.3 μmol/L、IBil 32.7 μmol/L。因胆红素下降欠佳,为进一步明确肝功能异常病因收入本科。既往史:有痛风病史4年多。其余无特殊。否认服药史、病毒性肝炎病史、血液病史,否认有毒有害物质接触史,无输血史,无手术、外伤史,育有一子,体健,无类似疾病病史,父母否认近亲婚育史。入院查体:神清,精神可,未见皮疹,未见肝掌及蜘蛛痣,浅表淋巴结未扪及肿大,双侧胸廓对称,双肺呼吸音清,未闻及干湿啰音。心律齐,心率74次/min,各瓣膜区未闻及病理性杂音。腹部平坦,腹软,无压痛、反跳痛及肌紧张,肝肋下未触及,脾脏肋下可触及,肋缘下约2.5 cm,移动性浊音阴性,肠鸣音4次/min,双下肢未见水肿。2021年2月18日血常规:WBC 5.04×109/L、HGB 122 g/L、PLT 104×109/L;肝功能:AST 60 U/L、ALT 62 U/L、ALP 157 U/L、GGT 49 U/L、TBA 119.2 μmol/L、TBil 287.2 μmol/L DBil 184.4 μmol/L、IBil 102.8 μmol/L。尿常规:尿胆原2+、胆红素3+。IgM 0.25 g/L、IgE 197 IU/mL,凝血无异常,甲、乙、丙、戊型肝炎病毒标志物均为阴性,自身免疫性肝病相关抗体(抗平滑肌抗体、抗肝/肾微粒体1型抗体、抗可溶性肝抗原/肝胰抗体、抗线粒体抗体M2亚型、抗Sp100、抗gp210抗体等)、铜蓝蛋白阴性。肿瘤标志物(AFP、CEA、CA125、CA19-9)均无异常。ERCP示(图1):胆总管胆泥。腹部增强CT示(图2):ERCP术后,肝管、胆管、胆囊内积气,胆囊壁积液,脾肿大、副脾,门静脉增粗。超声内镜示(图3):十二指肠乳头增大,胆总管下段截断性改变。因患者ERCP术后黄疸消退不明显,已排除自身免疫性肝病、肝外梗阻等一系列疾病,需考虑遗传性胆汁淤积性疾病可能。进一步完善肝组织活检示(图4):汇管区少量炎细胞浸润,肝细胞脂肪变性,点状坏死,毛细胆管扩张,淤胆,较多胆栓。免疫组化(因实验条件,未行MDR3免疫组化检查):CK7、CK19胆管上皮+,CK7阳性肝细胞约占10%;CD68示区域性活化的Kupffer细胞;α-SMA少部分活化肝星状细胞;MUM1少许浆细胞+;IgG个别+,IgG4-。Masson及天狼猩红染色示汇管区纤维组织增生,可见芒状纤维化及少数小叶间短纤维隔,轻度窦周纤维化,个别中央静脉内皮下纤维化;网染示肝板网状支架保存;PAS+,D-PAS示Kupffer细胞内蜡质样物,普鲁士蓝染色示肝细胞内少量铁颗粒沉积,铜染色-。结果提示:淤胆型肝炎,轻度炎症。需考虑家族性肝内胆汁淤积症。进一步行家族性胆汁淤积性基因检测,采用PCR+Sanger测序法检测。主要是通过高通量测序技术(二代测序)进行全外显子组测序检测:突变基因为ABCB4,染色体位置在chr7:87082270,突变信息为所编码的蛋白质第176位氨基酸残基由Arg突变为Gly。该变异为错义突变。结合患者病史、临床及体征、肝活检结果及基因检测,排除其他引起胆汁淤积的一系列疾病后,最终诊断:(1)PFIC3型;(2)胆汁淤积性肝炎;(3)慢性非萎缩性胃炎;(4)脾肿大。给予复方二氯酸醋酸二异丙胺保肝、丁二磺酸腺苷蛋氨酸退黄、雷贝拉唑护胃、UDCA 0.25 mg,3次/d口服。促进肝内胆汁排泄、补液等对症治疗,治疗好转后出院,出院口服UDCA治疗,患者2021年5月31日复查:AST 33 U/L、ALT 21 U/L、ALP 114 U/L、GGT 38 U/L、TBil 38.5 μmol/L、DBil 17.3 μmol/L、IBil 21.2 μmol/L。
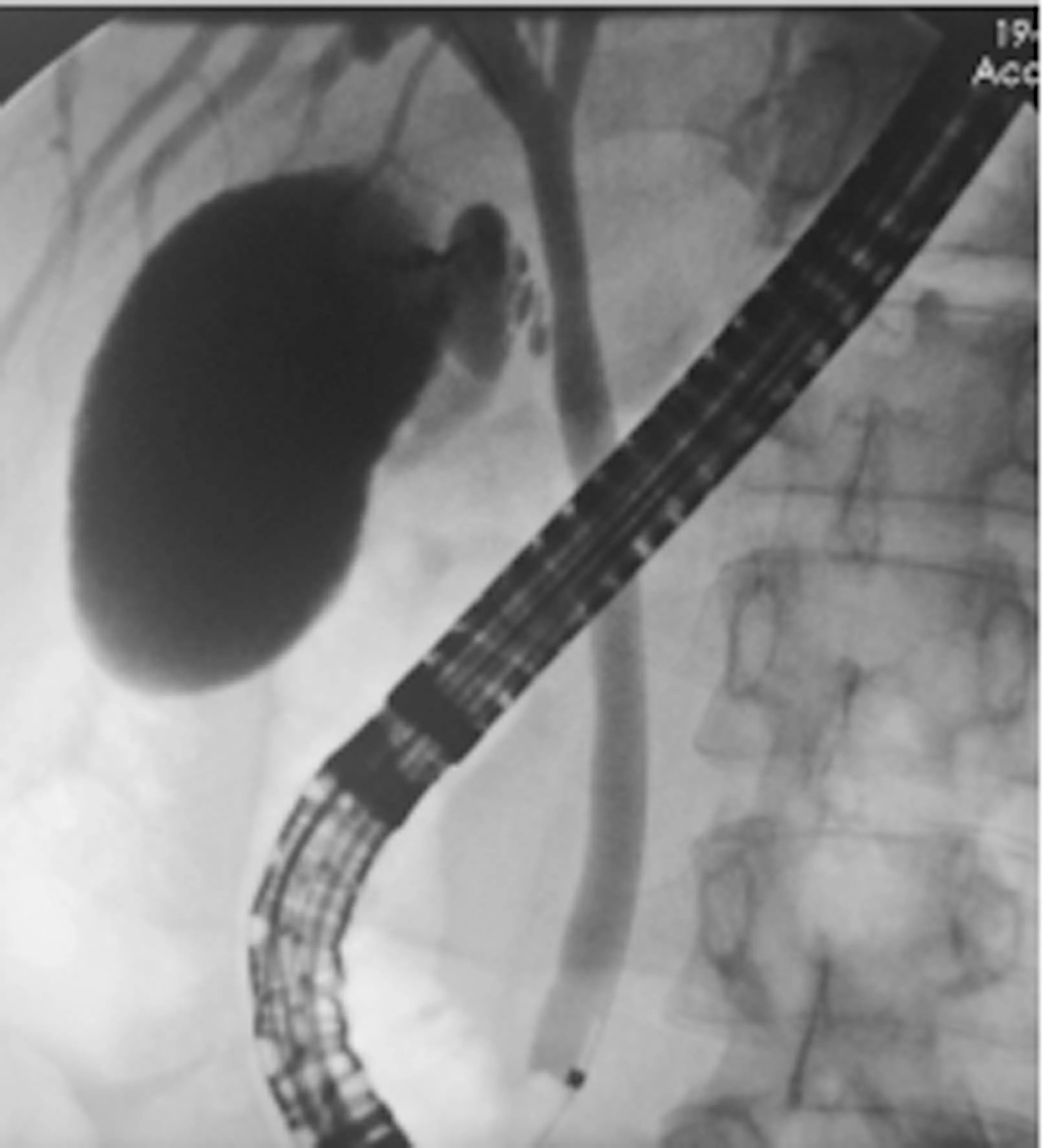
图1 ERCP示胆总管胆泥

注:a,胆囊壁积液,脾大;b,肝管、胆管内积气,门静脉增粗。
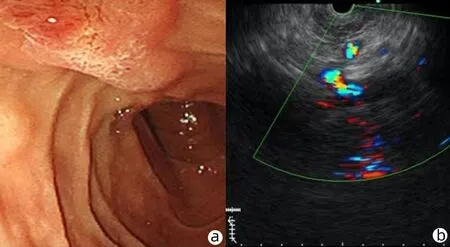
注:a,十二指肠乳头增大;b,胆总管下段截断性改变。

注:a,汇管区炎症细胞浸润(HE染色,×100);b,肝细胞脂肪变性,点状坏死,毛细胆管扩张,淤胆,较多胆栓(HE染色,×200);c,CK7胆管上皮(+)(免疫组化,×100);d,CD68活化的Kupffer细胞(免疫组化,×40);e,天狼猩红染色示汇管区纤维组织增生(×40);f,D-PAS示Kupffer细胞内蜡质样物(×100)。
2 讨论
临床上对于不明原因的胆汁淤积性肝病,以及血清GGT升高等,需考虑PFIC 3型可能,同时应排除其他原因所致的胆汁淤积,如病毒性肝炎、酒精性肝病、非酒精性脂肪性肝病、其他遗传性胆汁淤积性肝病、胆道闭锁、Alagille综合征、α1-抗胰蛋白酶缺乏症、囊性纤维化、硬化性胆管炎和肝内外梗阻等,并完善肝活检以及基因检测确诊[3]。
PFIC3是由ABCB4基因突变所致,ABCB4主要编码多耐药糖蛋白3型(multi-drug resistance 3,MDR3)基因,位于常染色体7q21区域,MDR3主要表达于肝细胞毛细胆管膜表面,将磷脂从肝细胞转运到胆管,同时与胆汁酸盐和胆固醇相结合,形成微粒[4],此时胆盐亲水性增加,去垢作用减弱,保护胆管细胞免受胆盐的毒性损伤,游离胆盐会直接对毛细胆管膜产生毒性的去垢作用,从而损伤胆管细胞,出现胆汁淤积、小胆管增生、炎症浸润,随后发生门管区纤维化、肝硬化。其中不稳定的混合微粒可胆固醇结晶,最终形成胆汁结石[5],形成的胆汁结石阻塞小胆道,而胆盐的分泌和排泄正常,大量胆盐导致GGT反流入血,导致血清GGT水平明显升高。目前已报道40多种不同的ABCB4基因突变。突变类型多为错义突变、无义突变、缺失和插入及剪切位点突变[6]。根据ABCB4基因突变致MDR3功能损害严重程度不同,可表现为PFIC3型或良性复发性肝内胆汁淤积症3型)。突变的程度决定了疾病的严重程度。PFIC3发病稍晚,少数在学龄期甚至青少年期起病,与其他几型PFIC相比,血生化GGT持续升高,AST/ALT轻度升高。在成年人,可表现为低磷脂性胆石症或妊娠时肝内胆汁淤积症。由于其胆汁中磷脂缺乏,皮肤瘙痒不重,肝组织病理特点:光镜下肝多核巨细胞样变、胆汁淤积、小胆管增生和门管区纤化、炎症浸润,晚期表现为肝硬化[7]。电镜下见胆固醇结晶。免疫组化提示小管MDR3蛋白缺失。
PFIC3治疗药物主要是UDCA[8],UDCA可改善50%PFIC3型患者的生化指标,长期服用UDCA可延缓肝硬化的进展。甚至某些病例可逆转纤维化。利福平可缓解瘙痒,晚期患者推荐肝移植治疗。该患者ERCP后复查胆红素下降欠佳,为明确病因收入本科,考虑胆红素下降欠佳原因:(1)患者术后各级胆管炎症等反应所致;(2)患者再次复查时,因UDCA治疗时间短,胆红素下降不明显,后续治疗后胆红素出现明显下降。
PFIC3 型为遗传性疾病,预后较差,对于GGT升高的不明原因的胆汁淤积以及合并胆石症形成的患者,需警惕此病,尽早完善基因检测、肝活检,完成早期诊断与早期治疗,可延缓疾病进展,提高生存率以及生活质量。同时家族一级亲属成员也应检测 ABCB4 基因突变情况以确定遗传概率,避免该病在家族中继续发生,该患者长期口服 UDCA后,血生化明显好转,需继续随访观察患者远期的发展。
伦理学声明:本例报告已获得患者及家属知情同意。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:张恬莹、周超、刘卫辉参与病史收集,撰写及修改论文;李贞茂、李良平、雷蕾负责拟定写作思路,指导撰写文章并最后定稿。
猜你喜欢
基层中医药(2022年7期)2022-11-17
理化检验(物理分册)(2022年7期)2022-08-03
保健医苑(2022年5期)2022-06-10
理论与改革(2021年5期)2021-09-16
中国生殖健康(2020年2期)2021-01-18
中国临床医学影像杂志(2019年6期)2019-08-27
——以咸阳市屯庄水库为例
水利科学与寒区工程(2019年2期)2019-04-28
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
中国神经精神疾病杂志(2014年1期)2014-03-01
