
高效液相色谱法测定曲伏噻吗滴眼液中马来酸噻吗洛尔及其有关物质含量
2022-11-15 08:06:18费路华费瀚雨吴娟
医药导报 2022年11期
费路华,费瀚雨,吴娟
(1.武汉药品医疗器械检验所,武汉 430075;2.华中科技大学同济医学院药学院,武汉 430030;3.武汉武药科技有限公司,武汉 430100)
曲伏噻吗滴眼液(商品名:苏力坦)原研厂家Alcon,2016年7月被国家药品审评中心纳入首仿品种实行优先审评评定名单。该药为复方制剂,每毫升溶液含曲伏前列素0.04 mg和马来酸噻吗洛尔5 mg(以噻吗洛尔计),主要用于降低成人开角型青光眼或高眼压症患者升高的眼压,适用于单用β-受体阻滞剂或前列腺素类似物局部治疗效果不佳者。
曲伏噻吗滴眼液在2020年版《中华人民共和国药典》及国外药典[英国药典(British Pharmacopoeia,BP)、美国药典(United States Pharmacopoeia,USP)、欧洲药典(European Pharmacopoeia,Ph.Eur )、日本药典(The Japanese Pharmacopoeia,JP)]中均未被收载,但《中华人民共和国药典》2020年版二部[1]、USP43版[2]及BP2021版[3-4]均收载有单方制剂马来酸塞吗洛尔滴眼液及其原料的质量标准,Ph.Eur(10.0版)[5]及JP(ⅩⅦ版)[6]中仅收载马来酸塞吗洛尔原料质量标准。USP43版马来酸塞吗洛尔原料的含量及有关物质均采用超高效液相色谱梯度法进行测定;滴眼液没有有关物质检查,含量测定用等度高效液相色谱(HPLC)法测定。BP(2021版)及Ph.Eur(10.0版)中马来酸塞吗洛尔原料质量控制方法完全一致,含量采用滴定法测定,有关物质检查采用HPLC梯度洗脱法;BP(2021版)中马来酸塞吗洛尔滴眼液含量采用提取分光法,有关物质采用HPLC等度分析方法。JP(ⅩⅦ版)含量采用滴定法,有关物质测定采用等度的HPLC法。《中华人民共和国药典》2020年版二部马来酸塞吗洛尔原料含量采用滴定法,有关物质采用薄层色谱法;滴眼液没有控制有关物质,含量采用紫外分光光度法。目前,国内外有文献报道[7-9]均为马来酸噻吗洛尔原料或滴眼液单方制剂,其含量、有关物质检查大多采用HPLC法,马来酸噻吗洛尔复方制剂质量控制笔者未见报道。笔者在本实验参照Ph.Eur(10.0版)优化色谱系统,建立曲伏噻吗滴眼液中马来酸噻吗洛尔含量及其有关物质测定方法。
1 仪器与试药
1.1仪器 Agilent1260高效液相色谱仪(G1311C四元泵、G1329B自动进样器、G1316A柱温箱、G1315D二极管阵列检测器、ChemStation工作站),XS105DU电子天平(瑞士Mettler Toledo公司,感量:0.01 mg),超纯水机(美国Millipore公司)。
1.2试药 马来酸噻吗洛尔对照品(USP对照品,批号:10k047,含量:99.9%)、噻吗洛尔系统适应性对照品(EP,批号:1.0,规格:每瓶5.045 mg,含马来酸噻吗洛尔及杂质B、C、D和F)、杂质D[4-(吗啉-4-基)-1,2,5-噻二唑-3-醇)(批号:1853)]、杂质G[4-(吗啉-4-基)-1,2,5-噻二唑-3(2H)酮-1-氧化物,批号:363950]、曲伏前列素(批号:110301,含量:98.6%)、曲伏噻吗滴眼液(规格:2.5 mL,批号:111201,111202,111203)均由武汉武药科技有限公司提供。甲醇和乙腈为色谱纯,水为超纯水,其他试剂均为分析纯。
2 方法与结果
2.1色谱条件 色谱柱为Agilent Zobax SB-C18柱(4.6 mm×250 mm,5 μm);以辛烷磺酸钠溶液(取辛烷磺酸钠2.16 g,加水700 mL溶解,加甲醇300 mL,混匀,冰醋酸调节pH值至3.0)为流动相A,甲醇为流动相B,梯度洗脱(0~20.0 min,90%A→40%A;20.0~30.0 min,40%A;30.0~31.0 min,40%A→90%A;31.0~41.0 min,90%A);检测波长:295 nm;柱温:30 ℃;流速:1.0 mL·min-1;进样量:20 μL。
取马来酸噻吗洛尔对照品2 mg,马来酸20 mg,加乙腈10 mL溶解,取1 mL在氮气流中吹干,在105 ℃干燥2 h,加流动相A 1 mL溶解,作为杂质E定位溶液;取马来酸噻吗洛尔系统适用性对照品1瓶,加流动相A 1 mL溶解,加杂质E定位溶液1 mL,摇匀,作为系统适用性溶液,量取20 μL注入液相色谱仪,记录色谱图,见图1。各相邻峰分离度均>1.5,符合要求。
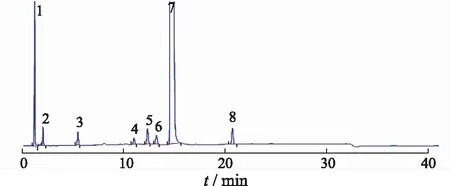
1.马来酸;2.杂质G;3.杂质D;4.杂质E;5.杂质F;6.杂质B;7.噻吗洛尔;8.杂质C。
2.2含量测定
2.2.1溶液的制备 对照品溶液:精密称取马来酸噻吗洛尔对照品约13.6 mg(约相当于噻吗洛尔10 mg),置100 mL量瓶,加水适量,超声使溶解,用水稀释至刻度,摇匀,即得。
供试品溶液:精密量取本品2 mL,置100 mL量瓶,用水稀释至刻度,摇匀,即得。
2.2.2线性关系考察 取马来酸噻吗洛尔对照品适量,加水制成每毫升含马来酸噻吗洛尔0.14,1.4,13.5,135.3,676.6及1 353.2 μg的溶液,按上述色谱条件测定,以浓度(C)为横坐标、峰面积(A)为纵坐标进行线性回归,得回归方程为:A=23.29C+18.12,r=1.000 0(n=6)。结果表明,马来酸噻吗洛尔在0.1~1353 μg· mL-1浓度范围内与峰面积线性关系良好。
2.2.3精密度实验 取同一份供试品溶液,按上述色谱条件连续进样6次,测定峰面积,峰面积RSD为0.07%(n=6),表明方法精密度良好。
2.2.4稳定性实验 取供试品溶液,分别于0,1,2,4,8,12,18 h按上述色谱条件进样测定,记录峰面积,结果马来酸噻吗洛尔的峰面积RSD为0.23%,表明溶液在室温下放置18 h内稳定。
2.2.5中间精密度实验 2名实验人员分别在HP1260高效液相色谱仪[色谱柱:资生堂ACR(4.6 mm×150 mm,5 μm)]、岛津LC-20A高效液相色谱仪[色谱柱:Waters XTerra®MS C18(4.6 mm×150 mm,5 μm)]上测定马来酸噻吗洛尔的含量,结果2人12份样品平均含量为101.1%,RSD为0.24%,表明方法重现性良好。
2.2.6回收率实验 按处方配制除马来酸噻吗洛尔之外的空白样品,取9份各2 mL,置100 mL量瓶,分别精密加入一定量马来酸噻吗洛尔,混匀,加水稀释至刻度,摇匀,即得回收样品溶液。按上述色谱条件测定,计算回收率。马来酸噻吗洛尔平均回收率101.1%,RSD=0.23%(n=9)。见表1。
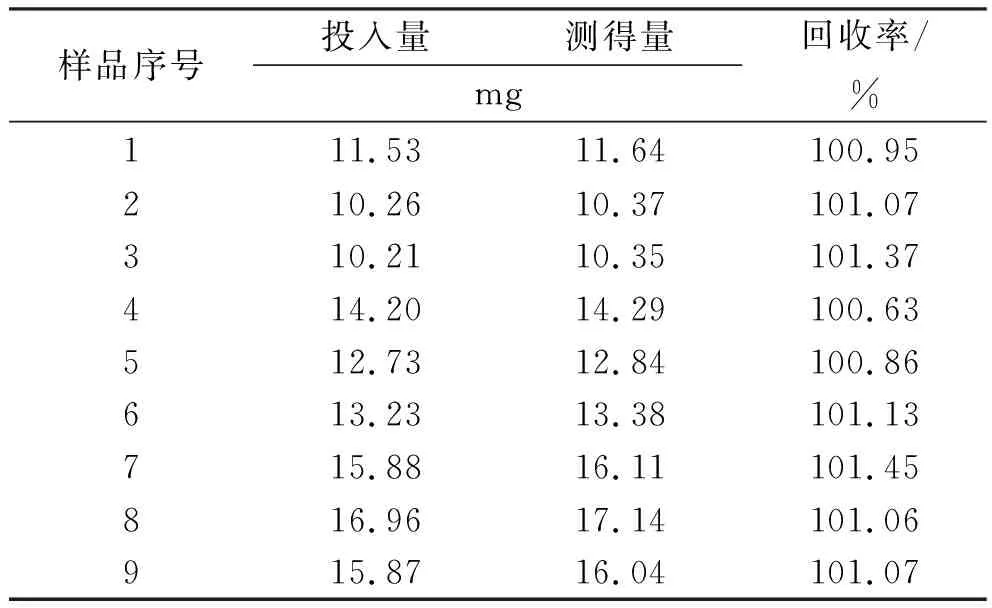
表1 马来酸噻吗洛尔回收率实验结果
2.2.7检测限 取“线性关系考察”项下溶液逐步稀释,按上述色谱条件进行测定。结果显示马来酸噻吗洛尔的检测限为1 ng(S/N=3),定量限为3 ng(S/N=10)。
2.2.8样品含量测定 精密吸取对照品溶液、供试品溶液各20 μL,按上述色谱条件进行测定;按外标法以峰面积计算,即得。每毫克马来酸噻吗洛尔相当于噻吗洛尔0.731 6 mg。结果见表2。
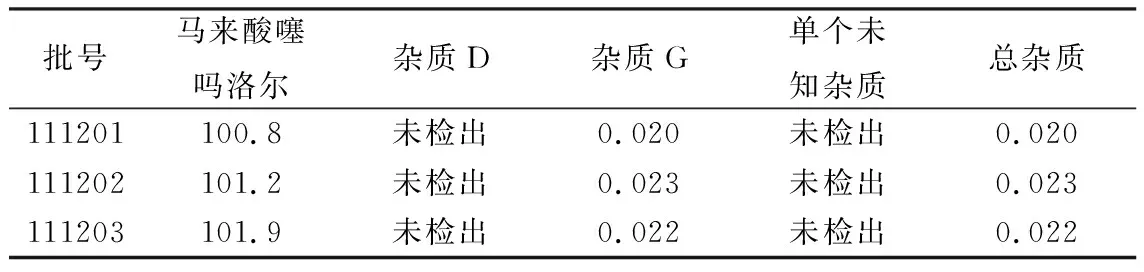
表2 样品含量测定结果
2.3有关物质测定
2.3.1溶液的制备 有关物质测定供试品溶液:取本品作为有关物质测定用供试品溶液。对照溶液:精密量取本品1 mL,置200 mL量瓶,加水稀释至刻度,摇匀,即得。空白辅料溶液:除马来酸噻吗洛尔外,其他成分按处方工艺配制即得。
2.3.2专属性实验 取马来酸噻吗洛尔6份,每份约70 mg,分别置10 mL量瓶,进行不同条件下的降解实验,实验条件分别如下,A.未经破坏;B.酸破坏:加3 mol·L-1盐酸5 mL,100 ℃破坏2 h后3 mol·L-1氢氧化钠溶液5 mL中和;C.碱破坏:加50%氢氧化钠溶液5 mL,100 ℃破坏30 min后用3 mol·L-1盐酸中和;D.热破坏:加流动相A 5 mL溶解,100 ℃加热破坏2 h;E.氧化破坏:加30%过氧化氢1 mL在室温下破坏60 min;F.光破坏:加流动相A 5 mL溶解,在365 nm波长紫外灯下光照12 h。
上述6种条件下的样品均用流动相A定容至刻度,摇匀,按上述色谱条件,分别进样,记录色谱图。实验表明:马来酸噻吗洛尔在各种破坏条件下均有不同程度的降解(图2),在酸、碱、加热、光照破坏条件下,均产生了杂质G;各降解产物均能与马来酸噻吗洛尔得到良好的分离,空白辅料溶液不干扰样品有关物质的测定。
2.3.3杂质线性关系考察 取马来酸噻吗洛尔杂质G和杂质D对照品适量,加甲醇-水(3:7)制成每毫升含马来酸噻吗洛尔杂质G和杂质D分别为 0.30,0.278;3.00,2.78;6.00,5.56,12.00,11.12;30.00,27.80;60.00,55.60 μg的系列溶液,按上述色谱条件进行测定,以浓度(μg· mL-1)为横坐标,峰面积为纵坐标进行线性回归,得回归方程,杂质G:A=27.39C+1.994 5,r=1.000 0;线性范围:0.3~60 μg· mL-1。杂质D:A=52.36C-6.074,r=1.000 0;线性范围:0.3~56 μg· mL-1。
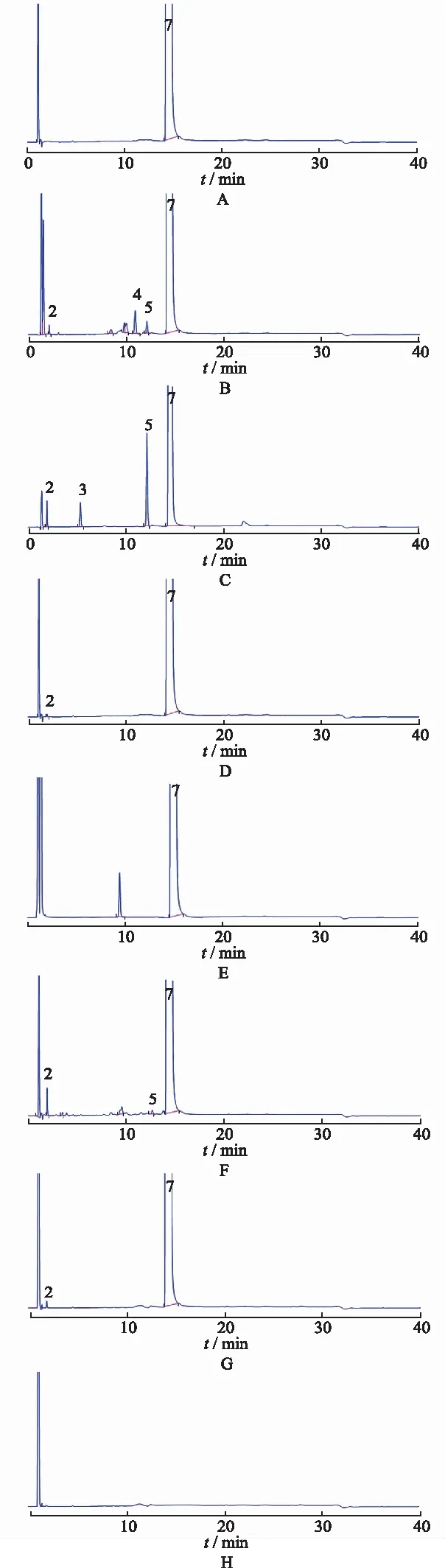
A.未破坏样品;B.酸破坏;C.碱破坏;D.热破坏;E.氧化破坏;F.光破坏;G.供试品溶液;H.空白辅料溶液;1.马来酸;2.杂质G;3.杂质D;4.杂质E;5.杂质F;6.杂质B;7.噻吗洛尔;8.杂质C。
2.3.4检测限 取杂质线性关系考察项下溶液逐步稀释,按上述色谱条件进行测定。结果按S/N=3计,杂质G和杂质D检出限均为0.3 ng。
2.3.5回收率实验 精密称取杂质D、杂质G各约10 mg,置同一200 mL量瓶,用流动相A溶解并稀释至刻度,摇匀,作为杂质回收实验储备溶液;精密量取5 mL,置25 mL量瓶,用水稀释至刻度,作为杂质回收实验的对照品溶液。称取未检出杂质D和杂质G的马来酸噻吗洛尔原料适量,按处方工艺制备曲伏噻吗滴眼液,作为杂质回收空白样品溶液。精密量取杂质回收实验的储备溶液3,5,8 mL(每个体积3份)分别置于25 mL量瓶,用杂质回收空白样品溶液稀释至刻度,摇匀,按上述色谱条件进行测定,按外标法以峰面积计算回收率,结果杂质D、杂质G回收率分别为101.3%(RSD=1.2%)、100.1%(RSD=1.3%)。
2.3.6校正因子实验 精密量取杂质回收实验储备溶液10 mL、对照品溶液5 mL置同一50 mL量瓶,用水稀释至刻度,按上述色谱条件进行测定,测得杂质D的校正因子为0.47,杂质G的校正因子为0.87。
2.3.7中间精密度实验 2名工作人员分别在HP1260高效液相色谱仪[色谱柱:资生堂ACR(4.6 mm×150 mm,5 μm)]、岛津LC-20A高效液相色谱仪[色谱柱:Waters XTerra®MS C18(4.6 mm×150 mm,5 μm)]上测定马来酸噻吗洛尔的有关物质,结果2人12份样品中均仅检出杂质G,平均含量0.02%,RSD 0.0%。
2.3.8耐用性实验 在上述色谱条件下,曾分别在3根不同的色谱柱[色谱柱:Ⅰ、资生堂ACR(4.6 mm×150 mm,5 μm),Ⅱ、Waters XTerra®MS C18(4.6 mm×150 mm,5 μm),Ⅲ、Agilent Zobax SB-C18(4.6 mm×250 mm,5 μm)]上进行耐用性实验;在同一色谱柱上[Agilent Zobax SB-C18(4.6 mm×250 mm,5 μm)]微调流动相的梯度比例,各杂质峰均能有效分离。
2.3.9有关物质检测 精密量取有关物质测定用供试品溶液、对照溶液各20 μL,按上述色谱条件进行测定,以加校正因子的主成分自身对照法计算各杂质含量,结果见表2。
3 讨论
3.1流动相的优化 用系统适用性溶液对流动相进行优化。采用Ph.Eur(10.0版)马来酸塞吗洛尔原料中有关物质分析方法不能有效分离杂质E和杂质F;随着甲醇浓度增加,各杂质可以有效分离,但杂质G保留时间较短,与溶剂峰不能很好分离;因此最终确定本方法流动相比例。在本流动相色谱体系,各杂质峰相对保留时间与Ph.Eur(10.0版)中相差较大,杂质B、杂质F和杂质C是根据Ph.Eur(10.0版)系统适用性溶液中出峰顺序定,杂质D和杂质G分别用其杂质对照品定位,杂质E定位溶液是按Ph.Eur(10.0版)中用于定位杂质E的方法配制的。
3.2测定波长的选择 曾尝试分别用220 nm(末端吸收)、276 nm(曲伏前列素的最大吸收波长)与295 nm(马来酸噻吗洛尔的最大吸收波长)作检测波长,结果发现在295 nm条件下,马来酸噻吗洛尔及其杂质峰的峰面积均大于其他波长,而且其他辅料及曲伏前列素没有干扰,故选择295 nm作为检测波长。
本品为复方制剂,有2个主药成分,一个是马来酸噻吗洛尔,另一个是曲伏前列素。在处方中马来酸噻吗洛尔的量是曲伏前列素的125倍,且曲伏前列素的最大吸收波长为276 nm,在295 nm波长处吸收值很小。在本文制订的色谱条件下,曲伏前列素主峰未能检测出,因此推测其有关物质就更加难以检测出;于是就判定未知的杂质峰均是由噻吗洛尔产生的。实验表明其他辅料均在马来酸之前出峰,不影响测定。由于杂质对照品较难获得,而且仅BP对马来酸噻吗洛尔滴眼液有有关物质控制,也是按未知杂质来控制的,因此我们暂参照其来制订标准,均作为未知杂质控制,以不加校正因子的主成分自身对照法进行计算,单个杂质不得过不得过0.4%,超过0.2%的杂质不得多于2个,总杂质不得过0.5%。
猜你喜欢
医学理论与实践(2023年18期)2023-10-24 06:40:19
中国典型病例大全(2022年13期)2022-05-10 23:54:31
中医眼耳鼻喉杂志(2021年1期)2021-07-22 07:38:10
蚌埠医学院学报(2020年11期)2020-12-17 05:16:20
中华养生保健(2020年2期)2020-11-16 00:49:10
中国卫生标准管理(2015年25期)2016-01-14 09:29:23
中国医疗美容(2015年5期)2015-02-03 03:02:01
中国药业(2014年19期)2014-05-17 03:12:22
济源职业技术学院学报(2014年3期)2014-02-28 02:35:42
中国现代应用药学(2013年4期)2013-03-11 19:13:53
