
The critical role of the endolysosomal system in cerebral ischemia
2022-11-11 10:28HuiYiZhangYeTianHanYanShiYaCaiYingXu
中国神经再生研究(英文版) 2023年5期
Hui-Yi Zhang, Ye Tian, Han-Yan Shi, Ya Cai, Ying Xu,
Abstract Cerebral ischemia is a serious disease that triggers sequential pathological mechanisms, leading to significant morbidity and mortality.Although most studies to date have typically focused on the lysosome, a single organelle, current evidence supports that the function of lysosomes cannot be separated from that of the endolysosomal system as a whole.The associated membrane fusion functions of this system play a crucial role in the biodegradation of cerebral ischemia-related products.Here, we review the regulation of and the changes that occur in the endolysosomal system after cerebral ischemia, focusing on the latest research progress on membrane fusion function.Numerous proteins, including N-ethylmaleimide-sensitive factor and lysosomal potassium channel transmembrane protein 175, regulate the function of this system.However, these proteins are abnormally expressed after cerebral ischemic injury, which disrupts the normal fusion function of membranes within the endolysosomal system and that between autophagosomes and lysosomes.This results in impaired “maturation” of the endolysosomal system and the collapse of energy metabolism balance and protein homeostasis maintained by the autophagy-lysosomal pathway.Autophagy is the final step in the endolysosomal pathway and contributes to maintaining the dynamic balance of the system.The process of autophagosome-lysosome fusion is a necessary part of autophagy and plays a crucial role in maintaining energy homeostasis and clearing aging proteins.We believe that,in cerebral ischemic injury, the endolysosomal system should be considered as a whole rather than focusing on the lysosome.Understanding how this dynamic system is regulated will provide new ideas for the treatment of cerebral ischemia.
Key Words: autophagy; biodegradation; brain injury; chaperone-mediated autophagy; endolysosomal system; fusion; hypoxia-ischemia, brain; mitophagy; N-ethylmaleimide-sensitive protein; TMEM175
Introduction
Cerebral ischemia is a common and serious life-threatening disease that is associated with high morbidity and mortality (Cramer et al., 2017).Brain injury after cerebral ischemia is a complex pathological process characterized by the interruption of cerebral blood flow and reperfusion, which can lead to irreversible neuronal damage, severe brain dysfunction, and longterm motor and cognitive impairment, and thus represents a risk factor for neurodegenerative disease (Kahl et al., 2018; Navarro Negredo et al., 2020).The main therapeutic goal in acute cerebral ischemia is to achieve vascular recanalization.Recombinant tissue plasminogen activator thrombolysis is the treatment of choice for acute cerebral ischemia (Mizuma et al.,2018).However, the application of thrombolysis is time-limited, and may further increase the ischemic area after blood recanalization (i.e., ischemiareperfusion injury) (Bhatia et al., 2010; Mizuma et al., 2018).After cerebral ischemic injury, numerous inflammatory factors, necrotic organelles, and neurodegenerative disease-related proteins accumulate in the brain,suggesting that neurons have a limited ability to clear and degrade harmful substances following cerebral ischemia (Kahl et al., 2018; Stoll and Nieswandt,2019).Improving the neuronal microenvironment after injury can contribute not only to the prevention of programmed cell death but also to the creation of conditions that favor neuronal repair and regeneration (Avraham et al.,2021).Accordingly, research attention has increasingly focused on the repair of the central nervous system, and particularly on the therapeutic potential of targeting lysosomes, the primary metabolite degradation machinery in the central nervous system (Zhang et al., 2021).
The robust degradative function of lysosomes is essential for maintaining both protein and cellular environmental homeostasis and lysosomal dysfunction can lead to the accumulation of intracellular waste, and even cell death (Hossain et al., 2021; Yang and Wang, 2021).Liu et al.(2019)found that lysosomal function differs in a cerebral ischemia stage-dependent manner.In the early stages of ischemia, the function of lysosomes is significantly enhanced, whereas, in the late stage of ischemia, lysosomal activity gradually declines, leading to protein accumulation and irreversible brain damage.Lysosomes are the terminal degradative compartment of autophagy, endocytosis, and phagocytosis, and can degrade a wide variety of waste, including damaged organelles and viral particles (Inpanathan and Botelho, 2019).Proteases are the predominant degradative molecules in lysosomes, and the ‘maturation’, function and loading of proteases in lysosomes are time-dependent (Repnik et al., 2013; Bright et al., 2016; Bissig et al., 2017).Accordingly, lysosomal function can have a significant influence on neuronal injury and repair after cerebral ischemia.We believe that the use of the term “lysosome” when discussing the impairment of degradation after cerebral ischemia is too limited.The term “endolysosomal system”more accurately reflects the dynamics and complexity of the system.The endolysosomal system generally refers to a series of organelles, including late endosomes, terminal lysosomes, and endolysosomes (Bright et al.,2016; Bissig et al., 2017).Late endosomes act as the stomach of the cells(Tjelle et al., 1996; Repnik et al., 2013), receiving degradative cargo from early endosomes, endolysosomal proteins from the Golgi apparatus, and waste from autophagosomes (Sahu et al., 2011; de Araujo et al., 2020).Late endosomes cannot effectively perform their degradative function owing to a high intraluminal PH and they must fuse with terminal lysosomes, yielding endolysosomes that have a lower PH, to achieve efficient degradation(Bright et al., 2016).Subsequently, endolysosomes are transformed into new terminal lysosomes, after which they fuse with late endosomes and enter the next round of endolysosomal cycling (Bright et al., 2016; Bissig et al., 2017;de Araujo et al., 2020).However, the exact biological processes performed by the endolysosomal system remain unclear.
Late endosomes are the precursors of endolysosomes and terminal lysosomes and contain similar protein components, such as lysosome-associated membrane protein 1/2 (LAMP-1/2) and cathepsin (Kuijpers et al., 2021).Studies involving lysosomes have largely used non-specific protein markers for their identification; accordingly, late endosomes and terminal lysosomes have often been confused (Lie and Nixon, 2019).Relatively recently, Vti1b,Rab7, and M6PR were found to be expressed in late endosomes, but not in terminal lysosomes.Thus, when using immunofluorescence staining, terminal lysosomes will be positive for LAMP-1/2 and cathepsins but negative for Vti1b, Rab7, and M6PR, potentially allowing to discriminate between the two organelles (Luzio et al., 2010; Kunwar et al., 2011; Yuan et al., 2018a).Electron microscopic analysis of late endosomes and terminal lysosomes has shown that the former contain an abundance of intraluminal vesicles and membrane fragments while the latter displays electron-dense regions with relatively few membrane fragments (Tjelle et al., 1996; Huotari and Helenius,2011).However, these features are not sufficient for effectively distinguishing the two organelles.
An important feature of the endolysosomal system is the acidic intraluminal environment on which the activity of many hydrolases is highly dependent (Xu and Ren, 2015).Terminal lysosomes contain only inactive acid hydrolases and can be recruited to the endolysosomal compartment and be re-synthesized from it, and they also serve as a reservoir of mature acid hydrolases (Bright et al., 2016).This feature can be used to distinguish endolysosomes from terminal lysosomes.The acidic environment of the endolysosomal system is maintained by multiple mechanisms, including via the pumping of protons into the lumen by vacuolar-type H-ATPases, which generates a large lumenpositive voltage (Ishida et al., 2013).In contrast to that proposed by Bright et al.(2016), namely, that the pH of membranes in the endolysosomal system is related to its location in the circulation, Johnson et al.(2016) suggested that the pH was instead dependent on the subcellular localization of the system.Disturbance of the acidic environment within the endolysosomal system leads to the release of cations into the cytoplasm, which disrupts intracellular ion homeostasis (Penny et al., 2015).Importantly, changes in pH affect fusion and hydrolytic functions in the endolysosomal system, resulting in the accumulation of undigested and harmful substances in the compartments,which impairs their normal physiological and biochemical functioning(Colacurcio and Nixon, 2016).Homeostasis among late endosomes, terminal lysosomes, and endolysosomes is critical for autophagy, proteostasis,ferroptosis, and other biological processes during cerebral ischemic injury, as well as for subsequent repair processes (Hu et al., 2021b).Cerebral ischemic injury affects the entire endolysosomal system, disrupts its dynamic balance,and has a serious impact on other physiological processes.
The above observations, together with recent studies showing that cerebral ischemia induces changes in the endolysosomal system, imply that a comprehensive understanding of the endolysosomal system in cerebral ischemia may contribute to the development of novel therapeutic strategies for this condition.Accordingly, in this review, we summarize current knowledge regarding the regulatory role of the endolysosomal system in cerebral ischemia.
Search Strategy
For this review, we searched PubMed for relevant studies published from 1990 to 2022.Broad text and MeSH terms were used to maximize sensitivity.The database search was conducted using a mixture of keyword-based and controlled vocabulary search strategies.Key search concepts were labeled as MeSH and were combined with key terms, as follows:
(1) (Hypoxia-Ischemia, Brain[Mesh] OR brain injury[tw]) AND (endolysosome system[tw] OR endolysosome*[tw]);
(2) (Hypoxia-Ischemia, Brain[Mesh] OR brain injury[tw]) AND(“N-Ethylmaleimide-Sensitive Protein”[Mesh] OR TMEM175[tw]);
(3) (Hypoxia-Ischemia, Brain[Mesh] OR brain injury[tw]) AND (Autophagy[Mesh] OR “Mitophagy”[Mesh] OR Chaperone-Mediated Autophagy[Mesh]).
The selected articles focused on the endolysosomal system and its membrane fusion function in cerebral ischemia.At least two independent researchers evaluated the reviewed articles and relevant information was extracted from every article.
The Regulation of the Endolysosomal System
The endolysosomal system is a highly ordered and dynamic system that involves, among other processes, late endosome fusion with terminal lysosomes and endosome fusion with cargos and autophagosomes, which contributes to maintaining intracellular protein homeostasis (Borchers et al., 2021).The system is regulated by a variety of protein complexes and membrane proteins; however, this regulation is altered during cerebral ischemic injury.
The regulation of NSF
Membrane trafficking involves membrane fusion between organelles or between vesicles and organelles (Yuan et al., 2018a).Membrane fusion in the endolysosomal system, including late endosome fusion with terminal lysosomes and terminal lysosome fusion with autolysosomes, is mediated by three core protein components—NSF, soluble NSF attachment protein(SNAP), and soluble NSF attachment protein receptors (SNAREs) (Bombardier and Munson, 2015; Yuan et al., 2018a).These components interact and correlate with each other, and coordinately regulate fusion processes in the endolysosomal system.
ATPases constitute one of the largest protein families and are known to participate in many complex biological processes.These proteins are characterized by the presence of an ATP enzymatic domain that drives conformational changes through ATP binding and hydrolysis (Ryu et al., 2016).The ATPase NSF, which functions as a homohexamer, is a classical type II AAAprotein containing a D1 domain that can reactivate SNARE complexes using energy derived from ATP hydrolysis (Yoon and Munson, 2018).Except for Drosophila, all organisms contain only one form of the NSF ATPase(Mohtashami et al., 2001), and inactivating NSF abolishes membrane fusion.SNAREs are fusion proteins and can be divided into vesicle SNAREs and target SNAREs (Liu et al., 2021).Interaction between the two types of SNAREs can result in their assembly into a core trans-SNARE that, in turn, mediates the fusion of two phospholipid membranes into an organelle (Hu et al., 2021b).After fusion, SNAREs form a stable complex that must be reactivated by NSF to participate in the next round of membrane fusion (Baker and Hughson,2016; Yoon and Munson, 2018).The interaction between NSF ATPase and SNAREs requires the adaptor protein SNAP (Hong and Lev, 2014).
Dalal et al.(2004) found that dominant interfering ATP hydrolysis-deficient NSF (E329Q) mutants accumulated in the Golgi and late endosomal membrane in mammalian cells, which led to a defect in membrane fusion.The authors further reported that the mutant ATPase was mainly deposited in the endolysosomal system, whereas relatively little NSF (E329Q) was recruited to the endoplasmic reticulum membrane (Dalal et al., 2004).The results of this study demonstrated that NSF-mediated membrane fusion plays an important role in the maintenance of the normal structure and function of the endolysosomal system, while other organelles are less dependent on this mechanism (Dalal et al., 2004).NSF-mediated fusion function merits further investigation given its reported importance for the endolysosomal system.
NSF-SNAP-SNARE machinery-mediated membrane fusion also requires the activity of other molecules, such as Rab GTPases and tethering complexes(Bröcker et al., 2010).They interact with each other and play a crucial role in membrane fusion events.In eukaryotes, tethering complexes can be further subdivided into two types, namely, homodimeric coiled-coil tethers and multi-subunit tethering complexes (Koike and Jahn, 2019).Early endosomal antigen 1 (EEA1) is a type of coiled-coil tether required for the fusion of early endosomes in eukaryotes (Das and Lambright, 2016).EEA1 consists of a FYVE (Fab1, YOTB, Vac1, EEA1) domain, two Rab5 interaction sites, and a long coiled-coil structure that is necessary for EEA1 aggregation (Bröcker et al., 2010).The C-terminal FYVE domain of EEA1 binds endosomal membranes containing phosphatidylinositol-3-phosphate (Larsen et al., 2021).Rab5 is a small GTPase, and the activity of PI3K is required for the fusion of Rab5-positive endosomal membranes and the creation of binding sites for EEA1 (Kamentseva et al., 2020).EEA1 acts as an interaction site for early endosomes and GTPases, thereby mediating the membrane fusion process in the endolysosomal system.
Class C core vacuole/endosome tethering (CORVET) and homotypic fusion and vacuole protein sorting (HOPS) are multi-subunit tethering complexes that play a prominent role in modulating membrane trafficking in endosomes and lysosomes (Nickerson et al., 2009).HOPS, a key molecule in late endosome-vacuole fusion, can recognize and interact with endolysosomal Rab7 (Plemel et al., 2011).HOPS may proofread trans-SNARE complexes and protect them from disassembly, thereby improving SNARE assembly efficiency(Lobingier and Merz, 2012; Lobingier et al., 2014).Almost all HOPS-related studies have been conducted in yeast; however, studies on animalsin vivo
have demonstrated that the function of HOPS as a tether in the process of lysosome fusion with late endosomes and autolysosomes is conserved across kingdoms (van der Kant et al., 2015; Wartosch et al., 2015).CORVET is thought to act upstream of HOPS in membrane fusion events involving the endolysosomal system (Peplowska et al., 2007).CORVET acts as a tethering complex that interacts with Vps21, a yeast homolog of Rab5 (Peplowska et al.,2007).However, the function of the CORVET complex in the endolysosomal trafficking pathway remains elusive and requires further investigation.The regulation of TMEM175
Using a cell patch clamp technique, Cang et al.(2015) identified a potassium channel, TMEM175, in lysosomes and late endosomes.This channel was subsequently found to consist of a complex of TMEM175 and protein kinase B(AKT) (Cang et al., 2015; Wie et al., 2021).Wie et al.(2021) further reported that TMEM175 is opened by conformational changes in AKT, but not its kinase activity.TMEM175 is essential for the regulation of lysosome-related properties, such as membrane potential, pH stability, and organelle fusion(Cang et al., 2015; Lee et al., 2017; Jinn et al., 2017; Zhang et al., 2020; Wie et al., 2021).The structure of TMEM175 is unique, consisting of two similar subunits composed of six transmembrane helices that form a tetrameric channel.Additionally, TMEM175 has no sequence homology to the tetrameric K+ channel and lacks a voltage-sensing domain and a P-loop selective filter found in the classical potassium channel (MacKinnon and Miller, 1989; Cang et al., 2015).TMEM175 maintains a potassium ion gradient across lysosomal and cytoplasmic membranes in a “leak-like” manner and stabilizes the resting potential (Cang et al., 2015).TMEM175 was once thought to exist only in prokaryotes but has since been detected in mammalian lysosomal membranes through proteomic analysis (Chapel et al., 2013).
Autophagosome-lysosome fusion has been reported to be accelerated in TMEM175-knockout RAW264.7 macrophages, while transfecting human TMEM175 into these macrophages partially restored the fusion rate (Cang et al., 2015).Jinn et al.(2017) and Wie et al.(2021) both reported that the loss of TMEM175 expression accelerated the fusion of autophagosomes with lysosomes in SH-SY5Y neuroblastoma cells and in TMEM175-knockout neurons, while Jinn et al.(2017) further indicated that autophagosome clearance was also affected.These results showed that TMEM175, via its role in regulating lysosomal fusion, performs a vital role in the maintenance of lysosomal function.As an important potassium channel in lysosomal membrane, the synergistic action with Na-K-ATPase and NSF will be an interesting research direction.
Several other categories of proteins are also proposed to mediate membrane fusion events.In mammalian cells, mitochondrial fusion is regulated by two types of mitochondria-related fusion proteins—mitofusin-1/2 (MFN1/2) and optic atrophy 1 (OPA1) (Chan, 2020)—both of which are members of the dynamin-related family of large GTP enzymes (Rovira-Llopis et al., 2017).MFN1 and MFN2 mainly mediate mitochondrial outer membrane fusionwhile OPA1 mediates mitochondrial inner membrane fusion (Giacomello et al., 2020).However, relatively few studies have focused on the function of these proteins in the endolysosomal system.Studies investigating protein–protein interactions will be important for the understanding of the molecular mechanisms involved in endolysosomal trafficking (Figure 1).

Figure 1| Schematic diagram of endolysosomal trafficking.
The Effect of Cerebral Ischemic Injury on the Endolysosomal System
Changes in NSF after cerebral ischemia
As mentioned above, there is only one type of NSF associated with membrane fusion events in mammalian cells (Mohtashami et al., 2001).Only cytoplasmic,free NSF has biological activity, and in a Triton-insoluble fraction, the activity of NSF is lost (Mohtashami et al., 2001; Sanyal and Krishnan, 2001).
Yuan et al.(2018b) established transient and global cerebral ischemia models in rats using bilateral common carotid artery ligation.The results showed that 20 minutes after the induction of transient cerebral ischemia, the NSF protein level was almost undetectable in the cytoplasm and was deposited in detergent salt-insoluble aggregates in hippocampal CA1 and Cx pyramidal neurons.In addition, NSF inactivation leads to the cessation of endolysosomal membrane transport and the impairment of endolysosomal fusion function,which further leads to neuronal damage after transient cerebral ischemia(Yuan et al., 2018b).Because excitatory pyramidal neurons contain numerous axonal endings and dendrites, they have a large membrane surface area, and the high concentration of NSF in pyramidal neurons may play a crucial role in maintaining neuronal excitability (Yuan et al., 2018b).After NSF inactivation,the endolysosomal membrane fusion dysfunction leads to membrane trafficking abnormalities.A large amount of 33-kDa cathepsin B (CTSB)accumulates in Golgi fragments, transport vesicles, and late endosomal structures (Yuan et al., 2018b).The CTSB leaking into the cytoplasm will further damage the endolysosomal system and lead to a cascade of events that results in irreversible damage (Yuan et al., 2018b).
In a subsequent experiment, Yuan et al.(2021) created a rat model of middle cerebral artery occlusion (MCAO) via right middle cerebral artery occlusion and obtained similar results.The levels of cytoplasmic, active NSF were gradually reduced from 1 hour of reperfusion until neuronal death 2 hours after MCAO.Furthermore, NSF inactivation mainly led to the interruption of late endosome fusion with lysosomes (Yuan et al., 2021).This is consistent with that previously reported by the same author, namely, cytoplasmic active NSF is the only ATPase responsible for the fusion of late endosomes with terminal lysosomes (Yuan et al., 2018b).The numbers of endosomes and autophagosomes, as well as protein levels, were reported to be increased in post-MCAO neurons, indicating that NSF inactivation blocks endolysosomal membrane trafficking and leads to endolysosomal structure aggregation (Yuan et al., 2021).In addition, early endosomes, autophagosomes, and protein aggregates are mainly concentrated in the cell body instead of distal synapses,which also supports that the cell body is the main region of endolysosomal degradation in the neuron.The main changes in penumbral neurons after focal cerebral ischemia include the irreversible fragmentation of the Golgi apparatus and the accumulation of CTSB-containing endolysosomes resulting from membrane fusion dysfunction, which is also consistent with previous studies (Yuan et al., 2021).The deposition or consumption of NSF, which plays a crucial role in the endolysosomal system, especially membrane fusion events, directly affects the efficiency of intracellular biodegradation during cerebral ischemic injury.
Changes in TMEM175 after cerebral ischemia
The potassium channel TMEM175 plays a key role in the regulation of lysosomal function, neuronal cell death, and neurological impairment after cerebral ischemia.Zhang et al.(2020) showed that the expression of TMEM175 in neurons was significantly downregulated after cerebral ischemia/reperfusion injury, decreasing progressively from 3 hours after injury and rebounding 12 hours later.In the same study, bothin vivo
andin vitro
experiments showed that decreased expression of TMEM175 damaged the acidic environment of the lysosome and affected the activity of lysosomal acidic hydrolases.The overexpression of TMEM175 through plasmid transfection significantly mitigated neuron death and behavioral deficits(Zhang et al., 2020).Notably, TMEM175 could reduce the accumulation of dysfunctional mitochondria, stabilize lysosomal pH, and restore the activity of acid hydrolases such as CTSB and cathepsin D.After cerebral ischemia, there is a correlation between neuronal damage in the hippocampal CA1 region and elevated levels of tau protein, indicating that cerebral ischemia may rely on tau-mediated neuronal death in the hippocampus (Pluta et al., 2018).Simultaneously, changes in the intrinsic environment, such as mitochondrial damage and excessive inflammatory cytokine production after cerebral ischemia, may be conducive to the accumulation of α-synuclein (Kim and Vemuganti, 2017).The accumulation of phosphorylated tau and abnormally folded α-synuclein are hallmark pathological features of Alzheimer’s disease (Bloom, 2014).In a survey of epidemiological risk factors for Alzheimer’s disease, Qiu et al.(2010) found that cerebral ischemia can initiate or accelerate neuronal degeneration.TMEM175 deficiency accelerates autophagosome-lysosome fusion, impairs autophagosome clearance, and increases the accumulation of phosphorylated α-synuclein in primary hippocampal neurons (Jinn et al., 2017).Compared with wild-type mice, the levels of phosphorylated α-synuclein (S129) were significantly increased in heterozygous TMEM175-knockout mice after exposing to α-synuclein preformed fibrils for 2 weeks, indicating that even a partial decrease in TMEM175 levels can affect lysosomal fusion function and aggravate neuronal damage (Wie et al., 2021).TMEM175 dysfunction plays a crucial role in the pathophysiology of cerebral ischemia and has been strongly implicated in the occurrence of neurodegenerative diseases, suggesting that TMEM175 may be a potential therapeutic target for neurodegenerative disease susceptibility resulting from cerebral ischemic injury.
Changes in cathepsin after cerebral ischemia
Yadati et al.(2020) found that cathepsin is the most abundant protease in the endolysosomal system.CTSB shows the highest levels of expression in neurons relative to other types of cathepsins (Petanceska et al., 1994).There are three types of CTSB, namely, a 46-kDa proCTSB form found in the Golgi, a 33-kDa CTSB present in late endosomes, and a 24–25 kDa CTSB found in lysosomes (Turk et al., 2012).The 46-kDa form is synthesized in the endoplasmic reticulum and transported via TVs to the Golgi apparatus;here, the TVs fuse with late endosomes, in which the 46-kDa CTSB is cleaved,yielding the 33-kDa CTSB; the late endosome then fuses with the terminal lysosome to form the endolysosome, where the 33-kDa CTSB is cleaved into the 24/25-kDa form (Pungercar et al., 2009; Huotari and Helenius, 2011; Turk et al., 2012).
The distribution of cathepsins in the endolysosomal system is altered after cerebral ischemia.Yuan et al.(2021) found that a large amount of CTSB accumulated in the damaged Golgi apparatus, while CTSB and endolysosomal structures wrapped with CTSB were seen to be dispersed in the cytoplasm and leaked out from damaged cells.In addition, the 33 kDa form of CTSB was found to be increased in the cytoplasm and endolysosomal compartments after cerebral ischemia, while the 46 kDa proCTSB and 24/25 kDa CTSB forms were correspondingly decreased (Yuan et al., 2018a).Moreover, using a rat model of hypoxia-ischemia, Troncoso et al.(2018) demonstrated that mature CTSD accumulated in the cytoplasm of cerebral cortex and hippocampal cells, indicating that these enzymes had been abnormally released from the lysosome to the cytoplasm.Prosaposin was observed to be similarly distributed.
Anagli et al.(2008) found that cysteine protease inhibitor-1 could mitigate the release of CTSB, neuronal death, and neurological impairment in the ischemic hemisphere of MCAO model rats.Meanwhile, Ni et al.(2015) reported that hypoxia/ischemia induced massive brain injury in wild-type neonatal mice,which could be partially attributed to the leakage of CTSB and cathepsin E following injury; however, brain insult was significantly less extensive in CTSBanimals.The extent and duration of cerebral ischemia injury differ in affected individuals, as does the amount of CTSB released.For instance,some studies reported that the structure of endolysosomes was only slightlydamaged and only a small amount of CTSB was released after transient cerebral ischemia, resulting in non-rupture cell death (Wang et al., 2018;Yuan et al., 2018a).In contrast, one study found that, after long-term global cerebral ischemic injury, a large amount of CTSB leaked into the cytoplasm and the extracellular space, resulting in the progression of brain injury (Yuan et al., 2021).Studies have shown that inhibiting cathepsin release can reduce neuronal death after cerebral ischemia.
CHNOmethyl ester (CA-074me) and aloxistatin (E64d) can suppress the release of CTSB after cerebral ischemia and relieve brain injury (Yamashima et al., 1998; Xu et al., 2016).However, studies have shown that CA-074me and E64d are not specific CTSB inhibitors (Montaser et al., 2002; Mihalik et al.,2004; Ryu et al., 2014).Indeed, CA-074me and E64d have been reported to significantly increase the half-life and concentration of CTSB, resulting in its delayed release and damage aggravation (Katunuma, 2010; Hook et al., 2014).In addition, both CA-074me and E64d are effective at the low pHs typically seen in the acidic lysosomal environment, but not under conditions of neutral pH (Cathers et al., 2002).These observations highlight the need to identify an inhibitor that has a strong inhibitory effect in the cytoplasm but not in the lysosome and one that can also specifically inhibit the release of CTSB after cerebral ischemia, thereby relieving neuronal injury.Such an inhibitor may also have potential as a therapeutic agent for cerebral ischemic injury.
Changes in autophagosome-lysosome fusion after cerebral ischemia Macroautophagy
Autophagy is a conservative biological process whereby damaged organelles and intracellular metabolic waste is degraded through the autophagylysosomal pathway (Klionsky et al., 2021).This biological process not only provides substances and energy for the body under stress but also helps maintain tissue homeostasis by digesting harmful substances (Kawabata and Yoshimori, 2020).There are three types of autophagy—macroautophagy,microautophagy, and chaperone-mediated autophagy (CMA) (Mizushima et al., 2011).The autophagy described in this section refers to macroautophagy.During autophagy, cargos are wrapped in a bi-layered membrane structure called an autophagosome, which fuses with lysosomes to form autolysosomes, in which the cargo is degraded (Klionsky, 2007; Mizushima,2007).
Autophagy is a complex biological process, and involves the nucleation,expansion, and closure of the autophagosome and its subsequent fusion with the lysosome, yielding the autolysosome (Lamb et al., 2013).The biodegradation process mainly occurs in autolysosomes (Lamb et al., 2013;Zhao and Zhang, 2019).Terminal lysosomes participate in the process of autophagy to maintain the dynamic balance of the endolysosomal system.The encounter of autophagosome and lysosome is the premise of fusion,which needs to be mediated by dynamic protein (Lőrincz and Juhász, 2020).The SNARE protein syntaxin 17, in conjunction with HOPS, plays a vital role in autophagosome-terminal lysosome fusion (Itakura et al., 2012).Tethering factors promote and stabilize the assembly of SNARE complexes and ensure that they are recruited to specific membranes to improve fusion efficiency (Yu and Hughson, 2010; Itakura et al., 2012).Many types of small GTPase, such as those belonging to the Rab and Ras families, act as molecular switches in the fusion process (Jackson and Bouvet, 2014; Lőrincz and Juhász, 2020).
Studies have shown that the autophagy-lysosome pathway is significantly inhibited during cerebral ischemic injury.Cheng et al.(2019) subjected SH-SY5Y neuroblastoma cells to oxygen-glucose deprivation (OGD) and observed that the co-localization of LC3 and LAMP-1 was decreased,indicating that autophagosome-lysosome fusion was impaired.In addition,sevoflurane postconditioning partially restored fusion function and provided neuroprotection (Cheng et al., 2019).In a rat model of chronic cerebral hypoperfusion, Che et al.(2017) found that autophagosome-lysosome fusion was decreased and further observed a large accumulation of autophagic vacuoles in primary hippocampal and cortical neurons.The inhibition of microRNA-27a expression can alleviate the damage due to autophagic vacuole accumulation by regulating LAMP-2 protein levels after cerebral ischemic injury (Che et al., 2017).Moreover, Zhou et al.(2017a) reported that OGD/R impaired lysosomal function and autophagy flux in hippocampal neurons in an oxygen-glucose deprivation/reperfusion (OGD/R) model,while mild hypothermia could regulate lysosomal function and increase autophagosome-lysosome fusion to protect neurons, effects that could be reversed by chloroquine treatment.However, some studies have also observed that cerebral ischemic injury can result in the excessive activation of the autophagy-lysosome pathway as well as increased neuronal injury (Zhou et al., 2017b; Shi et al., 2015; Tripathi et al., 2016).In a different study, Zhou et al.(2017b) established an OGD model in primary astrocytes and showed that the autophagy pathway was activated in model cells, which was involved in apoptosis via activating lysosomal protease, resulting in Bid cleavage and activating a series of caspase cascade.However, inhibiting autophagic activity stabilized the lysosomal membrane and protected cells from injury(Zhou et al., 2017b).Using anin vitro
model of OGD/R and anin vivo
model of transient global cerebral ischemia in rats, Chen et al.(2020) found that excessive autophagy induced by ischemic injury could lead to cell death, and blocking hyperpolarization-activated cyclic nucleotide-gated channel 2 (HCN2)protected neurons from OGD/R- and transient global cerebral ischemiainduced damage by accelerating autophagosome-lysosome fusion and autophagy degradation.The level of homocysteine is highly correlated with the degree of injury and mortality after cerebral ischemia (Shi et al., 2015).Tripathi et al.(2016) found that the use of homocysteine after OGD/R can inhibit autophagosome-lysosome fusion and lysosome degradation, resulting in increased cell death.Vitamin B supplementation can reduce cell death after stress, such as that induced by cerebral ischemia (Tripathi et al., 2016).The fusion of autophagosomes with terminal lysosomes is a key step in the late stage of autophagy.The role of the autophagy-lysosome pathway in cerebral ischemia merits further investigation (Table 1).Mitophagy
Mitophagy is generally considered to be a type of autophagy and the associated processes are similar (Swerdlow and Wilkins, 2020).Nevertheless,mitophagy has characteristic underlying mechanisms.The permeability of the mitochondrial membrane changes when mitochondria are damaged, which induces mitochondrial membrane depolarization and activates mitophagyrelated proteins (Belousov et al., 2021).Subsequently, the damaged mitochondria are wrapped in vesicles, forming phagosomes that are then transported to and fuse with lysosomes, where, finally, the mitochondria are degraded (Killackey et al., 2020).
A variety of signaling pathways regulate mitophagy, including the PTEN-induced kinase 1(PINK1)/Parkin pathway.Under steady-state conditions,PINK1 is localized to the cytoplasm and is transported to mitochondria where it is cleaved by mitochondrial processing peptidase (MPP) and presenilin-associated rhomboid-like protein (PARL) (Greene et al., 2012).Under pathological conditions, however, PINK1 cannot be transported to mitochondria and accumulates on the outer mitochondrial membrane(Yamano et al., 2016; Yan et al., 2020).Accumulated PINK1 recruits and activates Parkin, which initiates mitophagy, leading to the degradation of the affected mitochondria (Zhuang et al., 2016; Xu et al., 2021).Apelin-36, a neuroprotective peptide, protects HT22 cells from OGD/R-induced oxidative stress and mitochondrial dysfunction by promoting silent information regulator 2-related enzyme 1 (SIRT1)-mediated PINK1/Parkin-dependent mitophagy (Shao et al., 2021).Electroacupuncture can reduce the damage to mitochondrial function and the accumulation of damaged mitochondria induced by oxidative stress after cerebral ischemia through PINK1/Parkinmediated mitophagy, as well as exert a neuroprotective effect (Wang et al.,2019).Garciesculenxanthone B, a compound extracted from Geng Huang(Wu et al., 2021a), can induce the translocation of Parkin to damaged mitochondria, activate PINK1/Parkin-dependent mitophagy, inhibit apoptosis,reduce cerebral ischemia-reperfusion injury, and exert a protective effect on neurons following cerebral ischemia (Wu et al., 2021a).Hydrogen can protect mitochondrial function in injured hippocampal neurons after OGD/R by enhancing PINK1/Parkin-mediated mitophagy and can also protect cells from damage (Wu et al., 2018).Resveratrol can minimize ischemia-reperfusion injury and improve cell survival by activating PINK1/Parkin-mediated mitophagy in primary cortical neurons (Ye et al., 2021).It is essential to understand the role of the PINK1/Parkin signaling pathway in cerebral ischemic injury as regulating mitophagy via the PINK1-Parkin pathway may provide possible strategies for the treatment of cerebral ischemia injury (Table 2).
In addition to PINK1/Parkin, other signaling pathways and proteins are also known to regulate mitophagy (Table 3).These pathways/molecules can influence neuronal injury after cerebral ischemia by regulating mitochondria/lysosome fusion within the endolysosomal system.Mitophagy is considered to be a double-edged sword in cerebral ischemic injury.Some studies have shown that the activation of mitophagy has a protective effect on cerebral ischemic injury.The proteasome inhibitor carfilzomib reduces BCL2/adenovirus E1B interacting protein 3-like (BNIP3L) degradation by inhibiting the ubiquitin-proteasome pathway, rescues defective mitophagy, and reduces cerebral ischemic injury (Wu et al., 2021b).Esculetin can play a protective role in transient cerebral ischemia-reperfusion injury by reducing a series of oxidative stress cascades after cerebral ischemia, including increasing mitophagy and reducing mitochondrial damage (Xu et al., 2019).Studies bothin vivo
andin vitro
have shown that tissue plasminogen activator protects mitochondrial function through FUN14 domain-containing 1 (FUNDC1)-mediated mitophagy and reduces neuronal apoptosis induced by cerebral ischemia-reperfusion injury (Cai et al., 2021).Methylene blue was found to activate mitophagy through increasing mitochondrial membrane potential,reduce acute cerebral ischemia-induced brain injury, and ameliorate neurological function (Di et al., 2015).Methylene blue is also thought to hold promise as a neuroprotective drug in the treatment of acute ischemic stroke.Rapamycin, an activator of autophagy, can also enhance mitophagy by recruiting p62 to damaged mitochondria (Li et al., 2014).Rapamycininduced mitophagy accelerates the clearance of damaged mitochondria during cerebral ischemia and plays a protective role in the brain (Li et al.,2014).Some studies have indicated that excessive activation of mitophagy can increase neuronal death after cerebral ischemia, and inhibiting this overactivation can mitigate the induced brain damage.Salidroside recruits mechanistic target of rapamycin (mTOR) to mitochondria; activates the mTOR signaling pathway, thereby inhibiting excessive mitophagy; maintains the stability of mitochondrial quantity and function; and, ultimately, plays a neuroprotective role in OGD (Hu et al., 2021a).He et al.(2020) established a model of MACO in uncoupling protein 2 (UCP)-knockout mice and found that mitophagy and nerve injury were aggravated after cerebral ischemia in the animals, indicating that the overactivation of mitophagy has detrimental effects.Usingin vivo
andin vitro
models of cerebral ischemia-reperfusion injury, Zhang and Yu (2018) showed that nuclear receptor subfamily 4 group A member 1 (NR4A1) promotes cerebral ischemia-reperfusion injury by inhibiting MFN2-mediated mitophagy and blocking the MAPK-ERKCREB pathway and also alleviated cerebral ischemia-reperfusion injury by maintaining mitochondria homeostasis.
Table 1 |The role of the autophagy-lysosome pathway in cerebral ischemia
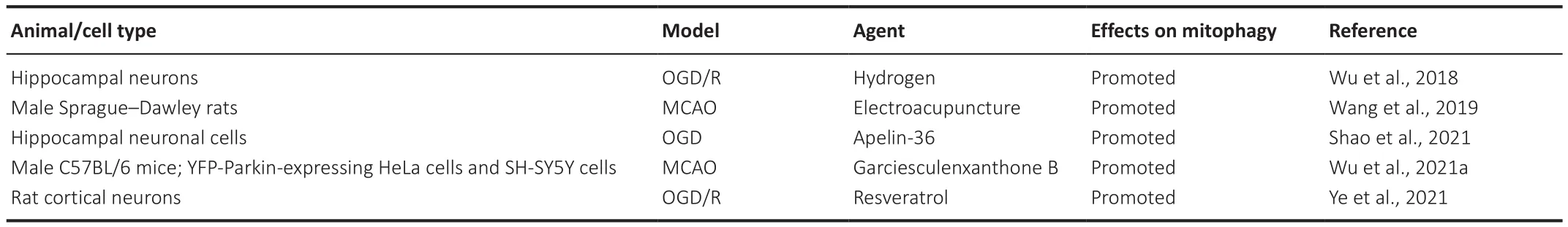
Table 2 |Agents regulating PINK1/Parkin-mediated mitophagy in cerebral ischemia

Table 3 |The role of mitophagy mediated by other pathways in cerebral ischemia
Chaperone-mediated autophagy
CMA is a form of lysosome-dependent autophagy that can selectively target proteins for degradation.In the initial stage, heat shock cognate protein 70(HSC70) binds to proteins with KFERQ-like motifs (Kirchner et al., 2019) in a manner that is independent of any specific property of the substrate protein(Bourdenx et al., 2021).LAMP-2A acts as a lysosomal receptor for CMA substrate proteins that must undergo unfolding before being transported into the lysosomal lumen (Bandyopadhyay and Cuervo, 2008).Helper proteins such as HSC70, HSP40, and HSC70-interacting protein are also involved in this process (Kaushik and Cuervo, 2018).CMA also involves the fusion of substrate proteins and lysosomes.LAMP-2 knockdown leads to a reduction in protein degradation in the heart, skeletal muscle, and other tissues where an extensive accumulation of autophagic vacuoles occurs (Tanaka et al., 2000).These observations indicate that LAMP-2 plays an indispensable role in the fusion of lysosomes and autophagic vacuoles.Lysosomal receptor proteins are synthesized in the rough endoplasmic reticulum, transported to the Golgi, sorted through endocytic pathways in early endosomes, and cycled in the endolysosomal system (Naslavsky and Caplan, 2018; Bajaj et al., 2019).However, little is known about the specific mechanisms underlying how the endolysosomal system affects CMA.
Dohi et al.(2012) showed that the expression level of LAMP-2A was increased in the ischemic hemisphere in anin vivo
model of cerebral ischemic injury,which indicated that CMA was activated after injury.They also observed similar resultsin vitro
, namely, CMA was found to be activated in Neuro2A cells under hypoxia, while siRNA-mediated knockdown of LAMP-2A led to apoptosis.Different from that macroautophagy was activated but did not promote cell survival, CMA can improve cell fate (Dohi et al., 2012).These results suggested that CMA may produce novel therapeutic targets for cerebral ischemia.An increasing number of studies have contributed to uncovering the role of CMA in regulating protein homeostasis and the progression of aging-related diseases such as Alzheimer’s disease and Parkinson’s disease.However, the pathological changes underlying Alzheimer’s disease after cerebral ischemia and the role of CMA in the development of injury require further study, as do the related molecular mechanisms.Conclusion and Perspectives
Cerebral ischemia is a serious, life-threatening cerebrovascular disease involving a variety of pathological factors, and can result in long-term learning and memory impairment (Fan et al., 2022; Peng et al., 2022; Yang et al., 2022).Given the limitations of current treatment strategies, more extensive studies on the pathophysiology of cerebral ischemia are needed.Lysosomes, the main degradative organelles in the central nervous system,play a critical role in the removal of damaged proteins, waste, and other organelles.Although studies have typically focused on the lysosome, a single organelle, the most recent data have demonstrated that the function of lysosomes cannot be separated from that of the endolysosomal system.The latter is a highly dynamic system comprising a series of dynamic, intertransforming organelles and involves different membrane fusion functions.Thus, “endolysosomal system” is a more accurate substitute for the term“lysosome”.
In this review, we focused on the endolysosomal system, especially its dynamic membrane fusion function, and discussed the changes in its function and regulatory factors during cerebral ischemia.NSF regulates endolysosomal trafficking pathways while the lysosomal potassium channel TMEM175 regulates the fusion between lysosomes and autophagosomes.Cerebral ischemia injury leads to aberrant expression and function of these proteins,which contribute to endolysosomal system dysfunction and neuronal cell death.The fusion of terminal lysosomes, which represent the final step of the endolysosomal system, with autophagosomes is a prerequisite for autophagy,a process that is critical for the maintenance of protein homeostasis.Regulating related proteins and biological processes may provide a potential therapeutic strategy for cerebral ischemic injury as well as an important experimental basis for the identification and development of therapeutic drugs and treatment strategies.However, the specific mechanisms involved in endolysosomal system trafficking remain unclear.
The regulation of proteins associated with endolysosomal system trafficking may lead to the identification of novel mechanisms that influence cerebral ischemia.Additionally, further studies are required on the interaction between these proteins and NSF, as well as on the influence of other lysosomal potassium channels on endolysosomal system-related fusion.Lysosomal BK channels also display high conductivity, and their activity is sufficient to induce significant changes in lysosomal membrane potential (Wang et al.,2017).However, whether BK channels also affect autophagosome-lysosome fusion remains unknown.LC3 tandem-tagged with mCherry and GFP can be used to observe the fusion of autophagosomes with lysosomes (Cang et al.,2015), and can also be used for related studies.Improvements in research methods targeting cerebral ischemia will likely contribute to improving long-term brain function after injury through the identification of the exact mechanisms involved in endolysosomal system trafficking and the influence of related potassium channels on fusion function.The development of effective therapeutics and successful clinical trials are also key goals.
There are still some limitations in this review.We only discussed a few major molecules that regulate endolysosomal trafficking, there are many other molecules involved in this process.Although significant progress has been made in understanding endolysosomal system in cerebral ischemia, many questions remain.For example, it remains unclear how endolysosomal system participates in other biological process after cerebral ischemia and the specific pathological mechanism involved in neuronal injury.Additionally, the potential differences between different degrees of cerebral ischemia increase the complexity of developing neural repair and regeneration targeting endolysosomal trafficking after cerebral ischemia.
In conclusion, numerous studies have contributed to the understanding of the mechanisms of injury and the pathological changes resulting from cerebral ischemia.The endolysosome system is thought to play a critical role in the maintenance of homeostasis after cerebral ischemia and may be a potential therapeutic target for this condition.Future studies focusing on the functional roles of the endolysosomal system as well as on the underlying mechanisms are needed to allow the development of targeted therapies for cerebral ischemia.
Author contributions:
YX, YT and HYZ conceptualized and designed the manuscript.HYZ drafted the manuscript, prepared the figure and conducted literature search.HYS and YC provided constructive suggestion and edited the manuscript.All authors approved the final version of the manuscript.
Conflicts of interest:
The authors declare no conflicts of interest.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
- 中国神经再生研究(英文版)的其它文章
- The effects and potential of microglial polarization and crosstalk with other cells of the central nervous system in the treatment of Alzheimer’s disease
- Blocking postsynaptic density-93 binding to C-X3-C motif chemokine ligand 1 promotes microglial phenotypic transformation during acute ischemic stroke
- Vimentin as a potential target for diverse nervous system diseases
- Clemastine in remyelination and protection of neurons and skeletal muscle after spinal cord injury
- Artificial nerve graft constructed by coculture of activated Schwann cells and human hair keratin for repair of peripheral nerve defects
- Novel therapeutic strategies targeting mitochondria as a gateway in neurodegeneration