
Activation of metabotropic glutamate receptor 1 regulates hippocampal CA1 region excitability in rats with status epilepticus by suppressing the HCN1 channel
2022-11-11 05:27XiaoDanLuoTaoXiangSiJunLiMeiGangMaMeiLingChenYuanWu
中国神经再生研究(英文版) 2023年3期
Xiao-Dan Luo, Tao Xiang, , Si-Jun Li, Mei-Gang Ma, Mei-Ling Chen, , Yuan Wu,
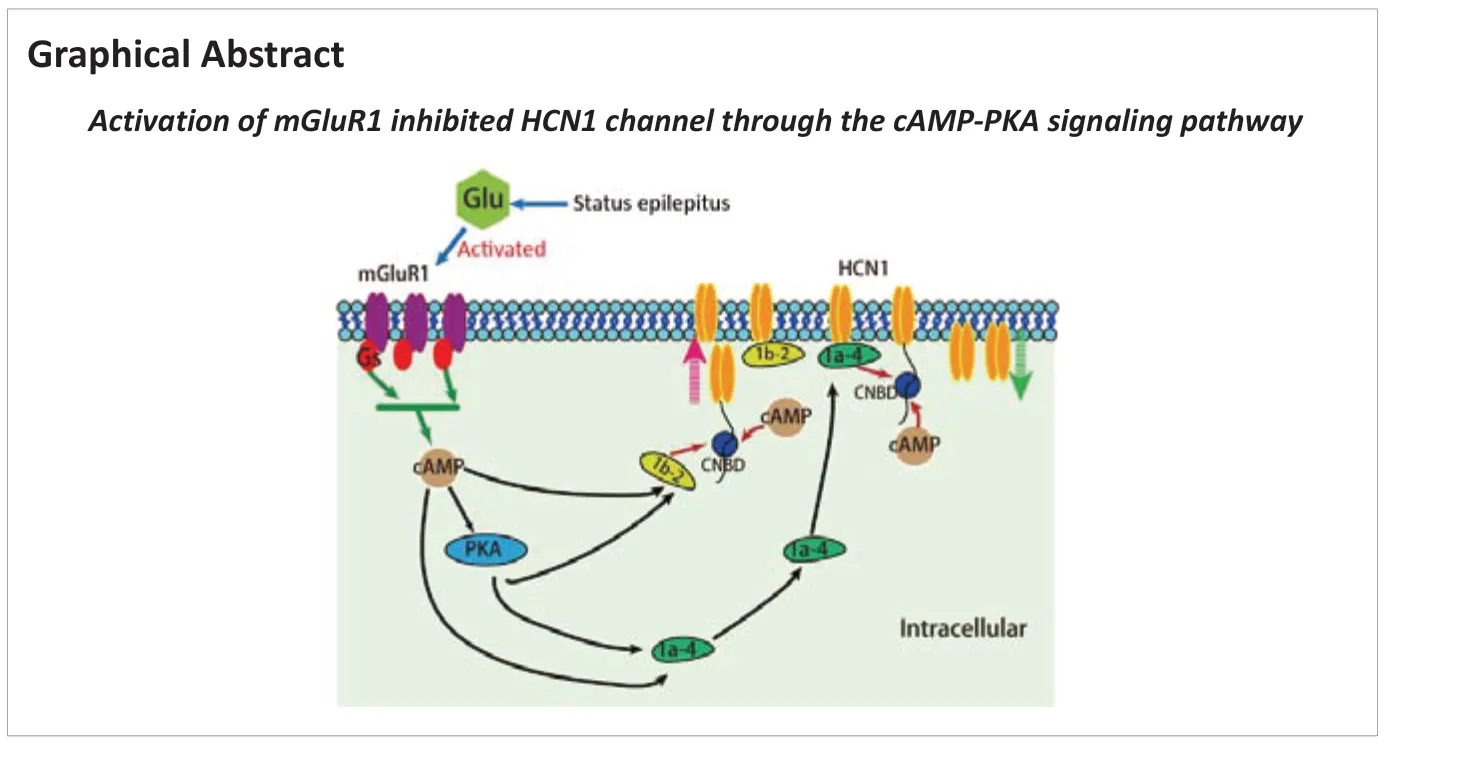
Abstract Dysregulation of hyperpolarization-activated cyclic nucleotide-gated cation (HCN) channels alters neuronal excitability. However, the role of HCN channels in status epilepticus is not fully understood. In this study, we established rat models of pentylenetetrazole-induced status epilepticus. We performed western blot assays and immunofluorescence staining. Our results showed that HCN1 channel protein expression, particularly HCN1 surface protein, was significantly decreased in the hippocampal CA1 region, whereas the expression of HCN2 channel protein was unchanged. Moreover, metabolic glutamate receptor 1 (mGluR1) protein expression was increased after status epilepticus. The mGluR1 agonist (RS)-3,5-dihydroxyphenylglycine injected intracerebroventricularly increased the sensitivity and severity of pentylenetetrazole-induced status epilepticus, whereas application of the mGluR1 antagonist (+)-2-methyl-4-carboxyphenylglycine (LY367385) alleviated the severity of pentylenetetrazole-induced status epilepticus. The results from double immunofluorescence labeling revealed that mGluR1 and HCN1 were co-localized in the CA1 region. Subsequently, a protein kinase A inhibitor (H89) administered intraperitoneally successfully reversed HCN1 channel inhibition, thereby suppressing the severity and prolonging the latency of pentylenetetrazole-induced status epilepticus. Furthermore, H89 reduced the level of mGluR1, downregulated cyclic adenosine monophosphate (cAMP)/protein kinase A expression, significantly increased tetratricopeptide repeat-containing Rab8b-interacting protein (TRIP8b) (1a-4) expression, and restored TRIP8b (1b-2) levels. TRIP8b (1a-4) and TRIP8b (1b-2) are subunits of Rab8b interacting protein that regulate HCN1 surface protein.
Key Words: (RS)-3,5-dihydroxyphenylglycine; CA1 region; excitability; H89; HCN1 channel; LY367385; mGluR1; pentylenetetrazole; status epilepticus
Introduction 594 Methods 595 Results 596 Discussion 598
Introduction
Status epilepticus (SE), which refers to repeated epileptic attacks that include unconscious intermissions or seizures sustained for more than 30 minutes (Weber et al., 2017), is a life-threatening emergency with a high mortality rate (Rosenow et al., 2007). SE causes brain damage through a series of cellular and molecular changes that eventually trigger spontaneous seizures (Postnikova et al., 2019; Liu et al., 2021); however, the exact pathogenesis of this disease remains unknown. Previous studies have revealed significant alterations in the function and expression of voltage-gated ion channels during SE. For example, Na, K, and Cavoltage-gated channels become dysfunctional during the pathogenesis of epilepsy (Depienne et al., 2009; Poolos and Johnston, 2012; Butler et al., 2018; Achar and Ghosh, 2021; Kinoshita and Koyama, 2021). Additional evidence has demonstrated the potential involvement of hyperpolarization-activated cyclic nucleotidegated cation (HCN) channels in epileptogenesis (Bonzanni et al., 2018; Lin et al., 2020). Specifically, HCN channels are voltage-gated ion channels highly expressed in the brain and associated with the onset of seizures (Jung et al., 2011). Results from animal experiments have revealed that HCN1 channel are strongly expressed in the hippocampus, superior colliculus, and cerebral cortex (Endo et al., 2008). HCN2 is extensively expressed in the brain cortex, whereas those for HCN3 mainly concentrated in the hypothalamus and HCN4 concentrated in thalamus (Notomi and Shigemoto, 2004).
Changes in HCN channels during SE have also been documented. For example, Powell et al. (2008) found that HCN channels were downregulated in the hippocampus in kainic acid (KA)-induced SE. Another study showed that HCN1 channel immunoreactivity was elevated in hippocampal pyramidal cell layers in a pilocarpine-induced SE model from 30 minutes to 12 hours after disease onset, whereas HCN2 immunoreactivity was suppressed in the dentate gyrus region (Oh et al., 2012). Results from a pilocarpine model revealed a reduction in HCN1 channel, whereas HCN2 channel remained unchanged (Santoro et al., 2010). Currently, there is limited information on HCN channels alterations during the early stages of SE. SE induced by KA and pilocarpine caused extensive neuronal death in the hippocampus at early stages (Curia et al., 2008; Richichi et al., 2008). However, in the early stages, seizures developed separately from neuronal death (Zhvania et al., 2015). Notably, pentylenetetrazole (PTZ)-induced SE recovered without spontaneous recurrence (Postnikova et al., 2019) and early neuronal death (Vasilev et al., 2018). Therefore, this model may be more suitable for studying the mechanism during the early stages of epilepsy (Zhvania et al., 2015).
Previous studies have shown that glutamate systems play an important role in the initiation and termination of SE (Devinsky et al., 2013; Furness et al., 2019; Green et al., 2021). Functionally, glutamate is released into synaptic gaps under physiological conditions and binds to glutamate receptors, leading to the propagation of motor potentials (Guo et al., 2010). Numerous studies have shown that metabolic glutamate receptors (mGluRs) are involved in the induction and maintenance of epilepsy (Ribeiro et al., 2010; Ghasemi et al., 2021). Particularly, the activation of group I mGluRs was found to increase epileptic activity (Keele et al., 1999), whereas exposure to group I mGluRs antagonists alleviated epileptic seizures, suggesting that group I mGluRs may be potential targets for antiepileptic therapy (Chapman et al., 1999; Shannon et al., 2005; Celli et al., 2019). mGluR1 and mGluR5 are two subtypes of group I mGluRs (Wierońska and Pilc, 2009). A previous study found that overexpressing mGluR1-enhanced green fluorescent protein in mice significantly increased the frequency of pilocarpine-induced SE (Pitsch et al., 2007), and mGluR1 upregulation was observed in epilepsy-associated focal cortical dysplasia (Aronica et al., 2003). Another study found that mGluR1-positive cells were distributed throughout the entire hippocampus (Guo et al., 2010), suggesting that mGluR1 may interact with HCN channels (Brager et al., 2012). mGluR1 activation in a rat model of chronic sciatic nerve constriction injury contributed to neuronal hyperexcitability by inhibiting HCN channels in the anterior cingulate cortex (Gao et al., 2016). To date, the potential roles of HCN channels and the regulation of mGluR1 and HCN channels in SE remain poorly understood.
Recent studies have provided evidence that tetratricopeptide repeatcontaining Rab8b-interacting protein (TRIP8b) is a regulatory subunit of HCN channels in the brain (Santoro et al., 2004; Lewis et al., 2009). In the hippocampus, TRIP8b is essential for maintaining the expression level and surface availability of HCN channels in the distal dendrites of pyramidal neurons (Lewis et al., 2011). Notably, TRIP8b (1a-4), TRIP8b (1a), and TRIP8b (1b-2) are the three predominantly expressed isoforms (Santoro et al., 2009). TRIP8b (1a-4) upregulates the surface expression of HCN1 (Santoro et al., 2009), whereas TRIP8b (1b-2) downregulates that of HCN1 and HCN2 (Santoro et al., 2004). Additional evidence has shown that TRIP8b interacts with the C-linker/cyclic nucleotide-binding domain (CNBD) of HCN1 channel (Han et al., 2011; Santoro et al., 2011). Because CNBD is also a binding site for cyclic adenosine monophosphate (cAMP), increased cAMP disrupts the interaction between TRIP8b and CNBD, thereby inhibiting the upregulation of HCN1 channel (Han et al., 2011). Previous studies have shown that mGluR1 couples to Gs protein, thereby increasing the cAMP concentration in certain cultured cells (Tateyama and Kubo, 2006; Cordeiro Matos et al., 2015). Moreover, mGluR1 mediates long-term potentiation in Purkinje neurons through the protein kinase A (PKA) signaling pathway (Sugiyama et al., 2008). Given that the actual role of these regulators (mGluR1, TRIP8b, cAMP, and PKA) in SE is currently unclear, we applied a widely used rat model of PTZ-induced SE to study HCN channels and elucidate the underlying molecular mechanisms in the hippocampal CA1 region during the early stages of SE development.
Methods
Animals
Because estrogen is a protective factor for epilepsy (Iacobaş et al., 2018), adult male Sprague-Dawley rats (aged 2‒3 months) with a body weight of 220‒250 g were purchased from the Experimental Animal Center, Guangxi Medical University, Nanning, China (Experiment project license No. SCXK (Gui) 2020-0003). The rats were habituated to standard conditions of 22 ± 2°C, 55 ± 5% humidity, and lighting conditions (a 12-hour light/dark schedule with lights on at 8:00 a.m.) for 7 days. All rats lived in clean cages and were allowed free access to food and water. The experiments were carried out between 9:00 a.m. and 5:00 p.m. All procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (8edition) (Yang et al., 2021). The experiments were approved by the Review Committee for the Care and Use of Laboratory Animals of Guangxi Medical University (experiment project license No. 202101010, approval date January 5, 2021). A minimum sample size of animals was calculated according to the results of preliminary experiments. All efforts were made to minimize the pain and suffering of animals. The rats were anesthetized with 3% sodium pentobarbital by intraperitoneal injection (30 mg/kg, P3761, Sigma-Aldrich) before decapitation.
Experimental design
Our study consisted of three experiments. The experimental design is shown inFigure 1
.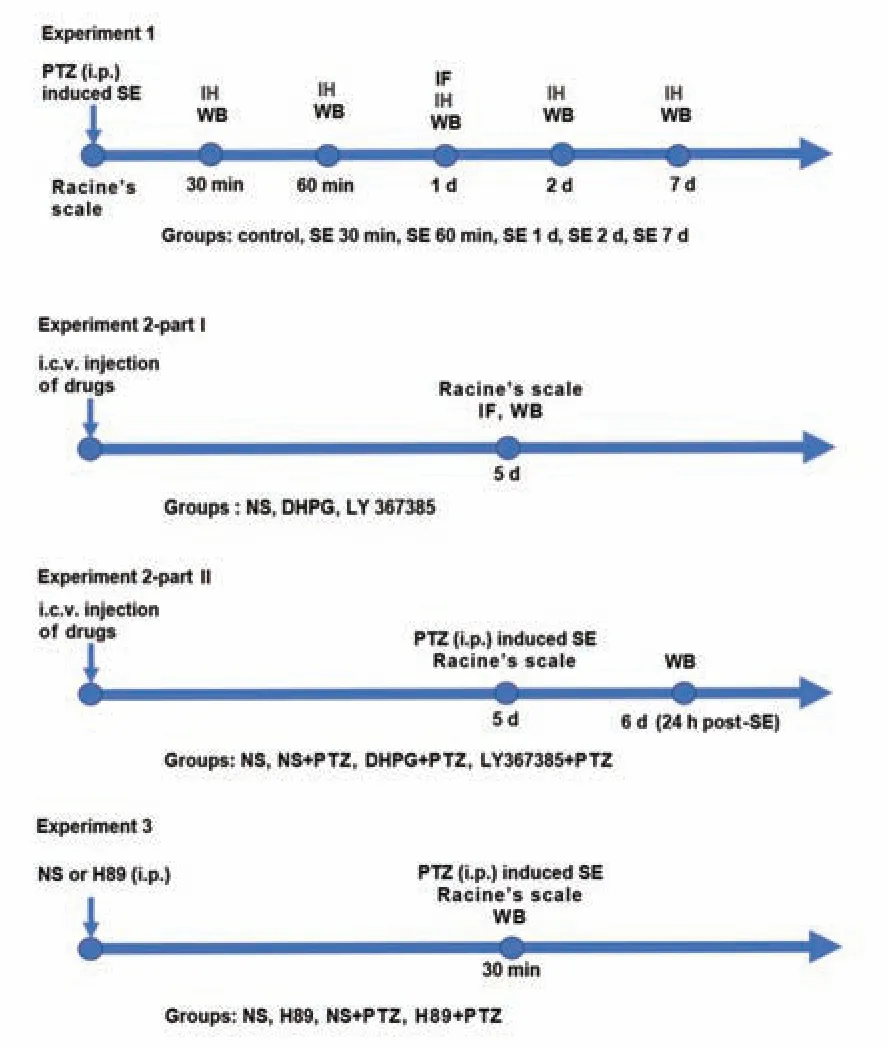
Figure 1 |Experimental design and animal groups.Experiment 1: Time course expression of HCN channels and mGluR1 after PTZ-induced SE; Experiment 2-part I: Clarification of the regulatory relationship between HCN1 and mGluR1; Experiment 2-part II: Investigation of the effect of an mGluR1 agonist and antagonist on SE; Experiment 3: Exploring the mechanism of mGluR1 activation and HCN1 inhibition. DHPG: (RS)-3,5-dihydroxyphenylglycine; HCN: hyperpolarized activated cyclic nucleotide-gated cation channels; i.c.v. injection: intracerebroventricular injection; i.p. injection: intraperitoneal injection; IF: immunofluorescence; IH: immunohistochemical staining; LY367385: (+)-2-methyl-4-carboxyphenylglycine; mGluR1: metabolic glutamate receptor 1; NS: normal saline; PTZ: pentylenetetrazole; SE: status epilepticus; WB: western blot.
Experiment 1
Ninety-six rats were randomly divided into a control group and 30 minutes, 60 minutes, 1 day, 2 days, and 7 days post-SE onset groups (n
= 16/group). Rats were first intraperitoneally injected with 55 mg/kg PTZ [10 mg/mL; P6500, Sigma-Aldrich, St. Louis, MO, USA; dissolved in normal saline (NS)], followed by 10 mg/kg every 5 minutes until SE occurred (Wang et al., 2017). This method reliably achieved a controllable intensity and sufficient duration of SE (approximately 30‒40 minutes). In the control group, rats were intraperitoneally injected with an equal volume (1.5‒2 mL) of NS.Using a modified Racine’s scale (Racine, 1972; Lüttjohann et al., 2009) to evaluate seizure phenotypes and stages, only rats with stage 4 and above were selected for this study. Seizure phenotypes and stages were determined as follows: stage 1, chewing or facial movement; stage 2, nod regularly; stage 3, unilateral or bilateral forelimb clonus; stage 4, bilateral limb clonus and rearing; and stage 5, generalized convulsions, falling, and rearing.
Experiment 2
For part I, 48 rats were assigned to sham, (RS)-3,5-dihydroxyphenylglycine (DHPG), and (+)-2-methyl-4-carboxyphenylglycine (LY367385) groups (n
= 16/group). DHPG (an agonist of group I mGluRs; single dose of 500 nmol/10 µL; HY-12598, MCE, Monmouth Junction, NJ, USA) (Ngomba et al., 2011) and LY367385 (a selective antagonist for mGluR1; single dose of 320 nmol/10 µL; HY-107515, MCE) (Chapman et al., 1999) dissolved in NS were injected into the right lateral ventricle [1.5‒2.0 mm right lateral to midline, 0.8‒1.0 mm posterior to bregma, 3.5‒4.0 mm ventral to the dura (Ni et al., 2020)] for 10 minutes using a 10-µL microsyringe (Gaoge, Shanghai, China) (Ni et al., 2020). In the control group, rats were intracerebroventricularly injected with 10 µL NS. Five days after recovery (Ni et al., 2020), spontaneous seizures were observed. Thereafter, rats were deeply anesthetized to collect brain samples.For part II, a mGluR1 agonist and mGluR1 antagonist were intracerebroventricularly injected to observe the effects of these drugs on excitability in PTZ-induced SE. Briefly, 68 rats were assigned to NS (n
= 8), NS + PTZ (n
= 20), DHPG + PTZ (n
= 20), and LY367385 + PTZ (n
= 20) groups. In part II, 10 µL of NS, DHPG, or LY367385 were intracerebroventricularly injected, and the rats were intraperitoneally administered PTZ as described in experiment 1 to induce SE. The behaviors of rats were observed, including the jerks latency, seizure scores (Racine’s scale), number of rats with SE, and mortality after SE. The jerks latency is the time from when PTZ (55 mg/kg) was first injected intraperitoneally to the induction of partial and/or generalized seizures (Zaitsev et al., 2015). At 24 hours post-SE induction, rats were deeply anesthetized to collect brain tissues.Experiment 3
Sixty-four rats were divided into NS, H89, NS + PTZ, and H89 + PTZ groups (n
= 16/group). All rats were intraperitoneally injected with NS (1.5‒2.0 mL) or H89 (a PKA inhibitor; dissolved in NS (Hosseini-Zare et al., 2011), 0.2 mg/100 g, HY-15979A, MCE). Rats in NS + PTZ or H89 + PTZ groups were intraperitoneally administered NS (1.5‒2.0 mL) or H89 30 minutes before treatment with PTZ as described in experiment 1. After 24 hours, all rats were deeply anesthetized to collect brain samples.Immunohistochemical and immunofluorescent staining
To observe changes in neuron and related protein expression in the hippocampal CA1 region after SE, immunohistochemical and immunofluorescence staining were performed. After being deeply anesthetized, rats underwent cardiac perfusion with NS, followed by 4% paraformaldehyde (G1101-500ML, Servicebio, Wuhan, China). The brains were collected and post-fixed in paraformaldehyde for 24 hours. Then, the brain samples were embedded in paraffin and cut into sections. Slides were dewaxed in xylene and dehydrated in graded ethanol solutions. The slides were boiled in sodium citrate buffer for 20 minutes to achieve antigen retrieval and then incubated with 3% hydrogen peroxide (180303, South Land Pharmaceutical, Guangdong, China) for 20 minutes and 5% bovine serum albumin (ST023, Beyotime, Shanghai, China) for 1 hour. The slides were incubated with rabbit anti-NeuN primary antibody (1:4000, Proteintech Group, Wuhan, China, Cat# 26975-1-AP) overnight at 4°C, washed, and incubated with horseradish peroxidase-conjugated anti-rabbit secondary antibody (1:200, Servicebio, Cat# GB23303) at 37°C for 1 hour. Subsequently, the slices were developed with a DAB Horseradish Peroxidase Color Development Kit (P0202, Beyotime) for 5 minutes, photographed, and analyzed using the Olympus Microscope System (Olympus, Tokyo, Japan). The number of NeuN-positive cells was calculated by ImageJ software (version 1.53f51; National Institutes of Health, Bethesda, MD, USA) (Schneider et al., 2012).
To investigate changes in the fluorescence intensity of HCN1 after SE, HCN1 immunofluorescence was performed. The slices were incubated with 10% goat serum (C0265, Beyotime) for 1 hour, followed by mouse anti-HCN1 primary antibody (1:200, Abcam, Cambridge, UK, Cat# ab84816, RRID: AB_2115171) at 4°C overnight. They were washed with phosphate-buffered saline and incubated with goat anti-rat IgG (1:200, Servicebio, Cat# GB21302) for 1 hour at 4°C, followed by counterstaining with 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (C1005, Beyotime).
For double immunofluorescence labeling of HCN1/mGluR1, the slices were incubated with a mixture of anti-HCN1 (mouse, monoclonal antibody, 1:200, Abcam, Cat# ab84816, RRID: AB_2115171) and anti-mGluR1 (rabbit, polyclonal antibody, 1:200, Abcam, Cat# ab82211) overnight at 4°C (Gao et al., 2016). Next, they were incubated with fluorescein isothiocyanate-conjugated goat anti-rabbit IgG (H+L) (1:200, Proteintech Group, Cat# SA00003-2, RRID: AB_2890897) and Cy3-conjugated goat anti-rat IgG (1:200; Servicebio, Cat# GB21302) for 1 hour at 4°C, counterstained with 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (C1005, Beyotime), photographed, and then analyzed using the Olympus Microscope System. Immunofluorescence intensity was calculated by ImageJ software using the following formula: average fluorescence intensity = total fluorescence intensity/area (Lin et al., 2020).
Western blot assay
Brains were rapidly harvested from anesthetized rats and submerged in ice-cold NS. The division of three regions of the hippocampus under a dissecting microscope (Li et al., 2014) is shown inFigure 2A
. Tissues of the hippocampal CA1 region were stored in an ultra-low temperature refrigerator at ‒80°C. Radio immunoprecipitation assay buffer (R0010, Solarbio, Beijing, China) containing protease inhibitors (A8260, Solarbio) was used to extract total protein. Membrane proteins were extracted using a Mem-PER Plus Membrane Protein Extraction Kit (89842, Thermo Scientific, Waltham, MA, USA). A BCA Protein Assay Kit (AR0146, Boster, Wuhan, China) was used to determine protein concentration. Samples (containing 30 µg of protein) were electrophoresed using 7.5‒10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels (PG111, PG112, Epizyme Biotech, Shanghai, China) and then transferred to polyvinylidene fluoride membranes (0.2 µm; Millipore, Darmstadt, Germany) for 30‒35 minutes in rapid transfer buffer (WB4600, NCM Biotech, Suzhou, China). Rapid blocking buffer (P30500, NCM Biotech) was used to block transferred membranes for 10 minutes at room temperature. Then, membranes were incubated with primary antibodies at 4°C overnight. Primary antibodies included mouse anti-HCN1 (1:1000, Abcam, Cat# ab84816, RRID: AB_2115171), mouse anti-HCN2 (1:1000, Abcam, Cat# ab84817, RRID: AB_1860532), rabbit anti-NeuN (1:1000, Proteintech Group, Cat# 26975-1-AP), rabbit anti-mGluR1 (1:500, Abcam, Cat# ab82211), mouse anti-TRIP8b (1a-4) (1:500, NeuroMab Clone, Los Angeles, CA, USA, Cat# N212/3), mouse anti-TRIP8b (1b-2) (1:200, NeuroMab Clone, Cat# N212A/34), rabbit anti-PKA α/β/γ (1:2500, Abcam, Cat# ab75991, RRID: AB_1524202), rabbit anti-β-tubulin antibody (1:1500, Cell Signaling Technology Inc., Danvers, MA, USA, Cat# 2128), and rabbit anti-ATPase alpha 1 subunit (ATP1A1 antibody (1:8000, Proteintech Group, Cat# 14418-1-AP). Goat anti-rabbit (1:5000, Proteintech Group, Cat# SA00001-2, RRID: AB_2722564) or goat anti-mouse (1:10,000, Cell Signaling Technology Inc., Cat# 5257) IgG H&L horseradish peroxidase was used as the secondary antibody. The membranes were scanned using an enhanced chemiluminescence system (ProteinSimple, Santa Clara, CA, USA). Total protein expression was normalized to the expression of β-tubulin, and the expression of surface proteins was normalized to the expression of ATP1A1 (Bakhache et al., 2020). Surface protein expression was normalized to total HCN1 levels in each sample lane. S/T is the ratio of (HCN1 surface expression/ATP1A1)/(HCN1 total protein expression/β-tubulin) (Song et al., 2014). A decrease in the S/T ratio is considered to indicate the internalization of membrane proteins, which are transported intracellularly (Jung et al., 2011). The decrease in S/T tends to cause neuronal excitation (Noam et al., 2010; Song et al., 2014; Mao and Wang, 2019). Therefore, we calculated the ratio of HCN1 S/T. All assays were performed at least three times.Measurement of cAMP concentrations
Tissues of the hippocampal CA1 region were rapidly resected and stored at ‒80°C until detection. A Rat cAMP ELISA Kit (JL10117, Jianglai Biotech, Shanghai, China) was used to measure cAMP concentrations. cAMP concentrations ranged between 0.375‒12 nM, and the coefficient of variation was 9‒11%. The absorbance was read at 450 nm using a spectrophotometer (Nanodrop, Thermo Scientific). The concentration was calculated according to the optical density using a standard curve.
Statistical analysis
Rats that died during the experiment and those with Racine’s scale scores less than stage 4 in SE groups were excluded from the experiment. The statistics of experimental data were calculated by an observer who was blinded to group allocations. Data are presented as the mean ± standard deviation and were analyzed with SPSS 26.0 (IBM, Armonk, NY, USA). Prism 5.0 (GraphPad Software Inc., San Diego, CA, USA) was used for mapping. Experimental data were compared between two groups using the Student’st
-test. Experimental data were compared among groups using one-way analysis of variance, followed by the least significant difference test.P
< 0.05 indicated statistical significance.Results
Time course expression of HCN channels after PTZ-induced SE
We monitored the progression of epileptic seizures after PTZ treatment, and seizures were not observed in the control group. We classified experimental rats according to Racine’s scale and found that all rats had SE after PTZ intervention. A small number of rats vomited pink foamy fluid during tonicclonic seizures, followed by the development of systemic cyanosis, respiratory arrest, and death. This PTZ-induced SE cohort displayed a mortality rate of 14% (11/80).
HCN channels consist of four subunits (HCN1‒4) (Berrera et al., 2006), among which, HCN1 and HCN2 represent the major subunits in the brain (Brewster et al., 2005). Here, we analyzed the expression of HCN1 and HCN2 proteins in the hippocampal CA1 region over time after SE using western blot assays. The results showed that HCN1 total protein was significantly downregulated at 1 day post-SE and was lower than that in the control group (P
< 0.001;Figure 2B
). There was no significant change in HCN2 total protein expression in CA1 regions between different times post-SE (P
> 0.05;Figure 2C
). These results were consistent with the findings of a previous study by Brennan et al. (2016). Immunofluorescence staining results revealed a significant decrease in HCN1 intensity at 1 day post-SE compared with that in the control group (P
< 0.01;Figure 2D
andE
). Collectively, these results suggested that HCN1 may play a crucial role in the mechanism underlying the pathophysiology of early-stage SE. Therefore, we further determined the change in HCN1 surface expression. The profiles of HCN1 surface expression in CA1 are shown inFigure 2F
. HCN1 surface expression rapidly decreased at 60 minutes post-SE (P
< 0.01) compared with that in the control group, decreased to the lowest point at 1 day post-SE (P
< 0.001), and then slightly but significantly recovered at 2 days post-SE (P
> 0.05). HCN1 surface expression was decreased at 1 day post-SE compared with that in the SE 60 minutes group (P
< 0.01;Figure 2F
). The ratio of HCN1 S/T was approximately 44.2 ± 3.2% in the control group (Figure 2G
). Notably, this ratio decreased at 60 minutes (P
< 0.05;Figure 2G
) and was the lowest at 1 day post-SE (P
< 0.001;Figure 2G
) compared with that in the control group. Consistent with HCN1 surface expression, the HCN1 S/T ratio was also decreased at 1 day post-SE compared with that in the SE 60 minutes group (P
< 0.05;Figure 2G
). Western blot analysis of surface protein extracts demonstrated no reactivity with anti-β-tubulin antibodies, indicating no cross-contamination of cytoplasmic fractions (Figure 2H
). Collectively, these results demonstrated the downregulation of total and surface HCN1 protein expression post-SE. However, HCN1 surface protein was downregulated at 60 minutes post-SE, earlier than HCN1 total protein. Moreover, the S/T ratio was lowest at 1 day post-SE.PTZ-induced SE does not cause neuronal death in the hippocampal CA1 region
A previous study showed that SE may cause neuronal death and subsequent neuronal loss (Yiş et al., 2013), processes that are also responsible for the suppression of HCN channels (Lin et al., 2020). In fact, the suppression of NeuN-positive neurons indicates the loss of neurons (Mullen et al., 1992; Vasilev et al., 2018). To explore whether PTZ-induced SE causes neuronal death or loss, we performed NeuN immunohistochemical staining of hippocampal sections followed by western blot assays in experiment 1. The results revealed the abundance of NeuN-positive neurons in the CA1 region of the hippocampus, with no statistically significant difference between the control and post-SE groups (P
> 0.05;Figure 3A
andB
). Similarly, we found no significant changes in NeuN protein expression between control and post-SE groups (P
> 0.05;Figure 3C
andD
). Taken together, these results indicated that PTZ-induced SE does not induce early neuronal loss in the hippocampal CA1 region, and suppression of the HCN1 channel post-SE is not caused by changes in the number of neurons.SE upregulates mGluR1 in the hippocampal CA1 region
Glutamatergic neurotransmission is considered an underlying factor of epilepsy (Guo et al., 2010), and endogenous glutamate is elevated after SE (Kanda et al., 1996). Most endogenous glutamate produced during epilepsy is removed through excitatory amino acid transporters (EAATs) and glutamate receptors (Guo et al., 2010). Notably, EAAT2 has been reported to be responsible for up to 90% of the total glutamate uptake (Tanaka et al., 1997), with the upregulation of EAAT2 protein shown to be a marker of increased glutamate concentrations (Vasilev et al., 2018). The results of experiment 1 showed that EAAT2 protein was significantly upregulated post-SE, specifically at 60 minutes (P
< 0.01), 1 day (P
< 0.01), and 2 days (P
< 0.05), and the degree of upregulation was higher than that in the control group (Figure 4A
andB
). Previous studies have shown that excess extracellular glutamate is an important risk factor for epilepsy as it over-activates glutamate receptors (Salt and Cordeiro, 2006; Seki and Lipton, 2008). In addition, evidence indicates an increase in glutamate binding to elevated group I mGluRs (mGluR1 and mGluR5) (Schröeder et al., 1999). In this study, we focused on mGluR1 expression owing to its relevance in the occurrence of epilepsy (Aronica et al., 2003). Our results showed that mGluR1 protein was significantly upregulated in the hippocampal CA1 region at 60 minutes post-SE (P
< 0.05) and increased to the highest level at 1 day (P
< 0.01) post-SE, followed by returning to control levels at 2 days (P
> 0.05;Figure 4A
andC
).mGluR1 and HCN1 are co-localized in the hippocampal CA1 region
The above results revealed upregulated mGluR1 protein and downregulated HCN1 protein at 1 day post-SE in the hippocampal CA1 region. This contrasting pattern of expression led us to hypothesize that the two proteins might be correlated. Previous studies have reported that HCN1 and mGluR1 are co-localized in the soma and apical dendrites of anterior cingulate cortex layer 5 pyramidal neurons (Gao et al., 2016). Therefore, we applied double immunofluorescence labeling to analyze their relationship in the hippocampal CA1 region in experiment 1. The results revealed that HCN1 and mGluR1 were co-localized in hippocampal CA1 pyramidal neurons (Figure 4D
).mGluR1 upregulates excitability in PTZ-induced SE by inhibiting HCN1 expression
Previous studies have shown that the activation of mGluRs potentially reduces HCN channels function by altering kinase activity and inducing entorhinal cortex plasticity (Brager and Johnston, 2007). The results of the present study revealed the upregulation of EAAT2, which subsequently activated mGluR1. Moreover, HCN1 channel protein was downregulated almost simultaneously, especially at 1 day post-SE. Double immunofluorescence labeling results in experiment 1 indicated that mGluR1 and HCN1 were colocalized in hippocampal CA1 pyramidal neurons. Thus, we hypothesized that increased glutamate after SE may activate mGluR1 expression, thereby inhibiting HCN1 channel to test this hypothesis, we evaluated the effect of mGluR1 agonist and antagonist on HCN1 channel. In part I of experiment 2, groups without PTZ administration showed no spontaneous epileptic seizures after the intracerebroventricular injection of drugs. Next, we evaluated changes in the expression of mGluR1 and HCN1 proteins in our model via western blot assays and immunofluorescence staining. The results revealed the significant downregulation of HCN1 total protein in the DHPG group (P
< 0.05), whereas mGluR1 protein was significantly upregulated in the DHPG group compared with that in the NS group (P
< 0.01;Figure 5A–C
). This effect simulated the alteration in HCN1 and mGluR1 protein at 1 day post-SE. These results imply that mGluR1 activation simultaneously upregulated mGluR1 and downregulated HCN1. Moreover, total HCN1 protein was slightly upregulated in the LY367385 group, although this was not significantly different compared with that in the NS group (P
> 0.05;Figure 5A–C
). Conversely, mGluR1 protein was significantly downregulated in the LY367385 compared with that in the control group (P
< 0.05;Figure 5A–C
). The results from double immunofluorescence labeling corroborated those from western blot assays (Figure 5D–F
). In part II of experiment 2, we tested the effects of mGluR1 agonist and antagonist on excitability by inducing SE with PTZ 5 days after the intracerebroventricular injection of drugs. The first PTZ injection (55 mg/kg, intraperitoneal) induced seizures in the NS + PTZ group (n
= 20), which were accompanied by a jerks latency of 1.38 ± 0.53 minutes (Zaitsev et al., 2015;Figure 6A
). The first PTZ injection-induced seizure scores ranged from 2 to 5 points in all tested animals, with an average of 3.5 ± 0.99 points (Figure
6B
). Subsequently, the number of intraperitoneal injections was selected according to the SE condition. All rats exhibited SE (Figure 6C
), with two of them dying, indicating a mortality rate of 10% (Figure 6D
). The latency of the jerks in the DHPG + PTZ group was 0.81 ± 0.34 minutes (Figure 6A
), and this was not significantly different from that in the NS + PTZ group. Rats in this group exhibited significantly higher seizure stages [increased to 4.2 ± 0.75 points] compared with those in the NS + PTZ group (P
< 0.05;Figure 6B
). Moreover, SE appeared in all rats, with a mortality rate of 40% (Figure 6D
). The latency of jerks in the LY367385 + PTZ group was 5.84 ± 1.77 minutes, which was significantly higher compared with that in the NS + PTZ group (P
< 0.001;Figure 6A
). The seizure stage for rats in this group decreased to 2.6 ± 0.68 points. Only nine (45%) rats developed SE (Figure 6C
), and no deaths were recorded (Figure 6D
).Western blot assays revealed significant upregulation of mGluR1 in the DHPG + PTZ group (P
< 0.05), whereas HCN1 total protein was significantly downregulated compared with that in the NS + PTZ group (P
< 0.01;Figure 6E–G
). In the LY367385 + PTZ group, mGluR1 protein was significantly downregulated after treatment with LY367385 and was significantly lower than that in the NS + PTZ group (P
< 0.05;Figure 6E
andF
), but not significantly different from that in the NS group (P
> 0.05). Moreover, HCN1 total protein was significantly upregulated in the LY367385 + PTZ group compared with that in the NS + PTZ group (P
< 0.01), but not the NS group (P
> 0.05;Figure 6E
andG
). Overall, these results indicated that DHPG activated mGluR1, inhibited HCN1 expression, and promoted SE progression. LY367385 exerted its antiepileptic effect by inhibiting the upregulation of mGluR1 and restoring the expression of HCN1 protein. The detection of HCN1 surface protein revealed that the degree of HCN1 surface protein downregulation was greater in the DHPG + PTZ group than in the NS + PTZ group (P
< 0.05;Figure 6E
andH
), although we found no significant difference in the S/T ratio between the two groups (P
> 0.05;Figure 6I
). In the LY367385 + PTZ group, HCN1 surface protein was significantly higher than that in NS + PTZ (P
< 0.001) and NS (P
< 0.001) groups (Figure 6H
). Similarly, the degree of increase in the S/T ratio in the LY367385 + PTZ group was greater than that in the NS + PTZ (P < 0.001) and NS (P < 0.01) groups (Figure 6I
). A previous study showed that the activation of mGluR1 may stimulate adenylate cyclase, which subsequently activates cAMP-dependent PKA (Schwartz, 1993). Because PKA is an upstream regulator of HCN1 protein (Shim et al., 2016), we investigated the effects of a mGluR1 agonist or mGluR1 antagonist on cAMP-PKA expression. The results revealed significantly higher cAMP concentrations (P
< 0.001;Figure 6J
) and PKA protein upregulation (P
< 0.001;Figure 6K
) in the NS + PTZ group compared with those in the NS group. These results were consistent with findings from a previous study (Hosseini-Zare et al., 2011). Notably, DHPG treatment significantly elevated the cAMP concentration (P
< 0.05;Figure 6J
) and upregulated PKA protein (P
< 0.001;Figure 6K
) in the DHPY + PTZ group compared with those in the NS + PTZ group. LY367385 treatment decreased the cAMP concentration (P
< 0.001;Figure 6J
) and downregulated PKA protein expression (P
< 0.001;Figure 6K
) compared with those in the NS + PTZ group. These results suggest that mGluR1 activation increases intracellular cAMP-PKA expression and downregulates HCN1 expression.cAMP-PKA regulates mGluR1 on HCN1 channel after SE
We explored the intracellular signaling pathway between mGluR1 activation and HCN1 downregulation after SE. The results from experiment 2 revealed that SE activated mGluR1, increased cAMP-PKA, and downregulated HCN1 protein. DHPG significantly elevated cAMP-PKA and downregulated HCN1 protein expression compared with NS + PTZ. Therefore, we hypothesized that the cAMP-PKA signaling pathway is a possible intracellular regulator of mGluR1 activation and HCN1 downregulation in SE. The results from experiment 3 showed that rats treated with H89 exhibited significantly longer jerks latency (P
< 0.001;Figure 7A
), but significantly lower seizure stages (P
< 0.05;Figure 7B
) compared with those in the NS + PTZ group. Only eight (50%) rats developed SE (Figure 7C
), and none of them died (Figure 7D
). Western blot assays revealed significant downregulation of mGluR1 in the H89 + PTZ group compared with that in the NS + PTZ group (P
< 0.05), but not the NS group (P
> 0.05;Figure 7E
andF
). Rats in the PTZ + H89 group exhibited significantly higher HCN1 total protein than that in the NS + PTZ group (P
< 0.01;Figure 7E
andG
) but not the NS group (P
> 0.05;Figure 7E
andG
). PKA was significantly downregulated in the PTZ + H89 group compared with that in the NS + PTZ group (P
< 0.05) but not the NS group (P
> 0.05;Figure 7E
andH
). We found no significant changes in the expression of mGluR1, HCN1 total protein, and PKA in the H89 group. HCN channels are modulated through the binding of cAMP to their CNBD (Saponaro et al., 2018). In addition, HCN channels are regulated by the auxiliary protein TRIP8b, which modulates trafficking and gating (Zolles et al., 2009). Moreover, TRIP8b is essential for maintaining the surface availability and expression levels of HCN1 channels (Lewis et al., 2011). To further explore the possible mechanism underlying mGluR1 and HCN1 channel function in SE, we detected the expression patterns of HCN1 surface protein and TRIP8b subunits (1a-4 and 1b-2) in the cAMP-PKA signaling pathway. The findings revealed that HCN1 surface protein was significantly downregulated in the NS + PTZ group compared with that in the NS group (P
< 0.01;Figure 7I
andJ
). HCN1 surface protein was significantly upregulated in the H89 + PTZ group compared with that in the NS + PTZ group (P
< 0.001;Figure 7I
andJ
). Moreover, HCN1 surface protein was significantly upregulated in the H89 + PTZ group compared with that in the NS group (P
< 0.001;Figure 7I
andJ
), and a similar trend was observed in the H89 group (P
< 0.001;Figure 7I
andJ
). Moreover, TRIP8b (1a-4) protein was significantly downregulated (P
< 0.01;Figure 7I
andK
), whereas TRIP8b (1b-2) protein was significantly upregulated (P < 0.001;Figure 7I
andL
) in the NS + PTZ group compared with those in the NS group. Furthermore, TRIP8b (1a-4) was significantly upregulated (P
< 0.001), whereas TRIP8b (1b-2) was significantly downregulated (P
< 0.001) in the H89 + PTZ group compared with those in the NS + PTZ group (Figure 7I
,K
, andL
). However, both were still higher than that in the NS group (TRIP8b (1a-4),P
< 0.05; TRIP8b (1b-2),P
< 0.001;Figure 7K
andL
). In the H89 group, TRIP8b (1a-4) protein was significantly upregulated (P
< 0.05), and TRIP8b (1b-2) remained unchanged (P
> 0.05) compared with those in the NS group (Figure 7I
,K
, andL
).
Figure 2 |Protein expression of HCN1 and HCN2 in the CA1 region after SE. (A) Schematic diagram showing the three regions of the hippocampus. The analyzed areas of the hippocampus (CA1, CA3, and DG) segmented by black lines. (B) Analysis of HCN1 levels in the hippocampal CA1 region after SE. The total protein level of HCN1 was downregulated at 1 day post-SE, as determined by western blot assays. (C) The total protein expression of HCN2 was not altered, as revealed by western blot analysis. (D) The immunofluorescence of HCN1 (red, Cy3, white arrow) in the hippocampal CA1 region. HCN1 fluorescence intensity was weakened at 1 day post-SE compared with that in the control group. Scale bars: 100 µm. (E) The mean fluorescence intensity of HCN1 in the SE 1 day group was significantly decreased compared with that in the control group. (F) The surface protein expression of HCN1 by western blot analysis. HCN1 surface expression was decreased at SE 60 minutes and the lowest at 1 day post-SE. (G) The ratio of HCN1 surface/total protein (S/T). Data are presented as the mean ± SD (n = 4/group). *P < 0.05, **P < 0.01, ***P < 0.001, vs. control group; #P < 0.05, ##P < 0.01, vs. SE 1 day group (one-way analysis of variance followed by the least significant difference test). ATP1A1: ATPase alpha 1 subunit; CA1: cornu ammonis 1; CA3: cornu ammonis 3; ctrl: control; DAPI: 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride; DG: dentate gyrus; HCN: hyperpolarized activated cyclic nucleotide-gated cation channels; SE: status epilepticus.
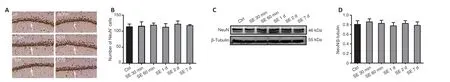
Figure 3 |NeuN-positive cells and NeuN protein in the hippocampal CA1 region after SE. (A) NeuN immunohistochemical staining (white arrow) in the hippocampal CA1 at different time points post-SE. NeuN-positive neurons (brown) exhibited no detectable cell loss. Scale bars: 50 µm. (B) The number of NeuN-positive cells in the hippocampal CA1 area. (C, D) Quantitative results of NeuN protein in the hippocampus using western blot analysis. Data are presented as the mean ± SD (n = 4/group) and were analyzed by one-way analysis of variance followed by the least significant difference post hoc test. CA1: Cornu ammonis 1; ctrl: control; HCN: hyperpolarized activated cyclic nucleotide-gated cation channels; NeuN: neuronal nuclei; SE: status epilepticus.

Figure 4 |Protein expression of EAAT2 and mGluR1 in the hippocampal CA1 region after SE. (A‒C) EAAT2 and mGluR1 expression were upregulated in the hippocampal CA1 region after SE, as determined using western blot analysis. Data are presented as the mean ± SD (n = 4/group). *P < 0.05, **P < 0.01, vs. control group (one-way analysis of variance followed by the least significant difference test). (D) Immunofluorescence images showing HCN1 (red, Cy3, white arrow) and mGluR1 (green, FITC, white arrow) co-localization in the hippocampal CA1 area. Scale bar: 20 µm. CA1: Cornu ammonis 1; ctrl: control; DAPI: 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride; EAAT2: excitatory amino acid transporter 2; HCN: hyperpolarized activated cyclic nucleotide-gated cation channels; mGluR1: metabolic glutamate receptor 1; SE: status epilepticus.

Figure 5 |Western blot analysis and immunofluorescence staining showing HCN1 and mGluR1 protein expression after SE in the presence of DHPG or LY367385. (A‒C) HCN1 was decreased, but mGluR1 protein was significantly increased in the DHPG group. In the LY group, HCN1 increased slightly but not significantly, whereas mGluR1 was significantly decreased. (D‒F) The mean fluorescence intensity of HCN1 (Cy3, red, white arrow) was decreased in the DHPG group. The mean fluorescence intensity of mGluR1 (green, FITC, white arrow) was increased in the DHPG group. In the LY group, the mean fluorescence intensity of HCN1 was unchanged, whereas that of mGluR1 was decreased. Scale bars: 20 µm. Data are presented as the mean ± SD (n = 4/group). *P < 0.05, **P < 0.01, vs. NS group (one-way analysis of variance followed by the least significant difference test). ctrl: Control; DHPG: (RS)-3,5-dihydroxyphenylglycine; HCN: hyperpolarized activated cyclic nucleotide-gated cation channels; i.c.v.: intracerebroventricular injection; LY: LY367385, (+)-2-methyl-4-carboxyphenylglycine; mGluR1: metabolic glutamate receptor 1; NS; normal saline; SE: status epilepticus.

Figure 6 |Activation of mGluR1 inhibits HCN1 channel through the cAMP-PKA pathway, increasing the epilepsy sensitivity and severity of PTZ-induced SE.(A) Graphs show the jerk latency (n = 20/group). (B) The average seizure score (n = 20/group). (C) Distribution of rats based on the number of SE. (D) Lethality (n = 20/group). (E‒H) Western blot results showing the expression of total HCN1, mGluR1, and surface HCN1 in the hippocampal CA1 region (n = 4/group). (I) The ratio of HCN1 surface/total protein (S/T). (J) The concentration of cAMP (n = 4/group). (K) Western blot results showing the expression of PKA (n = 4/group). Data are presented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, vs. NS group; #P < 0.05, ##P < 0.01, ###P < 0.001, vs. NS + PTZ group (one-way analysis of variance followed by the least significant difference test). ATP1A1: ATPase alpha 1 subunit; CA1: cornu ammonis 1; cAMP: cyclic adenosine monophosphate; DHPG: (RS)-3,5-dihydroxyphenylglycine; HCN: hyperpolarized activated cyclic nucleotide-gated cation channels; i.c.v.: intracerebroventricular injection; LY: (+)-2-methyl-4-carboxyphenylglycine; mGluR1: metabolic glutamate receptor 1; NS; normal saline; PKA: protein kinase A; PTZ: pentylenetetrazole; SE: status epilepticus.
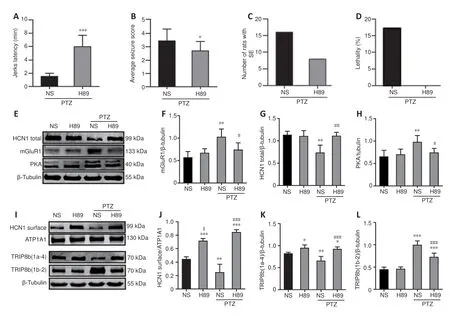
Figure 7 |H89 treatment decreases the severity of PTZ-induced SE. (A) Graphs show the jerk latency (n = 16/group). (B) Average seizure score (n = 16/group). (C) Distribution of rats based on the number of SE. (D) Treatment with H89 did not cause death in rats following PTZ-induced seizures (n = 16/group). (E‒H) Western blotting results showing the expression of total HCN1, mGluR1, and PKA. (I‒L) Surface expression of HCN1, TRIP8b (1a-4), and TRIP8b (1b-2) in the hippocampal CA1 region (n = 4/group). Data are presented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, vs. NS group; #P < 0.05, ###P < 0.001, vs. NS + PTZ group; $P < 0.05, vs. H89 + PTZ group (in behavioral results: the comparison between two groups was performed using the Student’s t-test; others: one-way analysis of variance followed by the least significant difference test). CA1: Cornu ammonis 1; cAMP: cyclic adenosine monophosphate; H89: PKA inhibitor; HCN: hyperpolarized activated cyclic nucleotide-gated cation channels; mGluR1: metabolic glutamate receptor 1; NS; normal saline; PKA: protein kinase A; PTZ: pentylenetetrazole; SE: status epilepticus.
Discussion
Previous studies have shown that SE may induce changes in synaptic properties and intrinsic membrane characteristics (Beck and Yaari, 2008), neurogenesis (Pitkänen and Lukasiuk, 2009), the extracellular matrix (Smirnova et al., 2018), and axonal sprouting and dendritic remodeling (Amakhin et al., 2017). However, data on HCN channels alterations during the early stage of SE remain limited. Findings from epileptic animal models have shown that the CA1 region of the hippocampus is one of the key areas regulating epilepsy in the brain (Curia et al., 2008; Lüttjohann et al., 2009). Although several factors have been implicated in SE development, we mainly focused on the role of HCN channels, which have been shown to mediate theI
current (Bonzanni et al., 2018). There are two different mechanisms through whichI
affects the excitability of neurons. TheI
current can be deactivated by depolarization (Marcelin et al., 2009) and activated by hyperpolarization of the cell membrane, a phenomenon that endows theI
current with unique and multiple functions to regulate neuronal excitability (Robinson and Siegelbaum, 2003). Notably, different brain regions and various cell types have distinct neuron excitabilities because of differences in the subcellular distribution of HCN channels (Santoro and Baram, 2003). Studies have shown that most HCN1 and HCN2 channels are distributed in the distal dendrites of hippocampal pyramidal neurons, where they regulate resting membrane potential (Ludwig et al., 2003; Santoro et al., 2010; Kozák, 2019).The first research showing changes in HCN channels expression during epileptogenesis described febrile seizures, which chronically enhancedI
in CA1 pyramidal neurons and significantly slowed the activation and deactivation kinetics (Chen et al., 2001). The expression profiles of HCN channels following febrile and KA-induced seizures revealed persistent loss of HCN1 mRNA and protein expression, as well as transient downregulation of HCN2 expression (Brewster et al., 2002, 2005). Moreover, Powell et al. (2008) revealed significant downregulation of HCN1 and HCN2 mRNAs in the CA1 regions and entorhinal cortex of a KA-induced model at 24 hours, 7 days, and 6 weeks after SE. Moreover, HCN1 mRNA was downregulated after a brief increase post-SE in the hippocampal CA3 region and dentate gyrus (Powell et al., 2008). Later, Jung et al. (2011) found a reduction in HCN1 protein in the CA1 region in pilocarpine-induced SE. Neuronal cell loss in the hippocampus is a significant feature of KA- or pilocarpine-induced seizure models (Morimoto et al., 2004). Neuronal loss may contribute to the decrease in HCN channels expression (Powell et al., 2008). Studies have confirmed that KA- or pilocarpine-induced SE models show extensive neuronal death in the early stage (Ye et al., 2020; Xiang et al., 2021). Recent studies on the ultrastructure of neurons showed that PTZ-induced SE resulted in instantaneous mass atrophy, high alkaline, and the generation of high electron density (dark) cells but did not lead to neuronal cell death (Noam et al., 2010; Zaitsev et al., 2015; Zhvania et al., 2015). Our results were consistent with the previous findings that revealed no significant changes in the number of NeuN-positive neurons and alterations in NeuN protein in the hippocampal CA1 area in rats with PTZinduced SE. Thus, the changes in HCN channels in this model can be observed more intuitively.The results of the present study indicated that HCN1 total protein was significantly downregulated in the hippocampal CA1 region of rats at 1 day post-SE, consistent with the findings from a previous report (Jung et al., 2011). We found no changes in the expression of HCN2 protein; thus, we concluded that changes in HCN1 protein play a more important role in the pathogenesis of epilepsy. Previous studies have shown that although the sensitivity of HCN1 to cAMP is not as high as that of HCN2 (Möller et al., 2014), HCN1 responds faster to cAMP than HCN2 (Li et al., 2014). The activation of HCN2 channel is slower during the neonatal stage, but the rapid activation and cAMP insensitivity of HCN1 channel occur in the mature stage (Brewster et al., 2007). This suggests that the downregulation of HCN1 channel protein is more likely to cause neuronal excitability in the early stage of SE, similar to that in the immature brain (Lin et al., 2020).
Loss of theI
current increases the intrinsic excitability of neurons and contributes to the occurrence of epilepsy (Dyhrfjeld-Johnsen et al., 2008), and the intrinsic excitability of neurons depends on cell membrane ion channels (Santoro et al., 2010). In the present study, we detected changes in the expression of HCN1 surface proteins in the CA1 region after SE. Results from experiment 1 showed that, in the CA1 region of the control group, HCN1 protein expressed on the cell surface accounted for approximately 44.2% of the total protein, which was less than the 75% previously reported by Jung et al. (2011). This difference may be related to the different collection methods of hippocampal CA1 specimens. We used the entire CA1 region brain tissue for protein extraction, whereas Jung et al. (2011) used electrophysiological hippocampal slices for the microdissection of the CA1 region. The brain tissue they collected may contain pure CA1 pyramidal neurons. In addition, HCN1 surface protein and S/T ratios began to decrease 60 minutes after SE, but HCN1 total protein was downregulated at 1 day post-SE, which may indicate that the rapid internalization of surface protein occurred at 60 minutes post-SE. The downregulation of HCN1 surface protein and reduction in the S/T ratio may downregulate functional proteins on the cell surface, thereby increasing the intrinsic excitability of neurons (Jung et al., 2011; Lin et al., 2020). The S/T ratio was the lowest at 1 day post-SE, indicating marked membrane internalization (Mao and Wang, 2019). Our results on the intrinsic excitability of neurons were consistent with those of Postnikova et al. (2019), who found that the hippocampal neurons became more excitable and started firing at a reduced excitatory input because of a significant increase in Ri at 1 day post-SE in the single PTZ-induced model using a patch clamp. PTZ treatment caused rapid SE development, which was spontaneously relieved. However, the detection of HCN1 surface protein and total protein in our study and the electrophysiological detection by Postnikova et al. (2019) showed that the excitability of neurons was the highest at 1 day post-SE. Although there were no seizures at this time, neuronal excitability remained high. This phenomenon caused us to speculate whether it is necessary to administer treatment to reduce neuron excitability even though there were no seizures and requires further exploration. HCN1 total protein decreased to the lowest level at 1 day post-SE, which was consistent with the results of a study by Jung et al. (2011). Consequently, we speculated that SE rapidly induced the internalization of HCN1 surface protein, followed by its degradation at 1 day post-SE (Jung et al., 2011). Although HCN1 total protein had not recovered back to control levels at 2 days post-SE, the surface protein and S/T ratio were restored, suggesting that neuronal excitability recovered back to the baseline level. Notably, the downregulation of HCN1 channel occurred extremely rapidly in PTZ-induced SE rats. Therefore, elucidating the underlying mechanisms may provide relevant insight into the early processes of epilepsy development. The induction and maintenance of elevated neuronal excitability mediated by group I mGluRs are considered key steps in epilepsy development (Chapman et al., 1999; Shannon et al., 2005). In addition, the antiepileptic effect of mGluR1 receptor antagonists is considered a possible target for the treatment of epilepsy (Shannon et al., 2005). HCN1 is highly expressed in the hippocampus (Endo et al., 2008), similar to mGluR1 (Ngomba et al., 2011), and HCN1 and mGluR1 co-localize in the anterior cingulate cortex of the cerebral cortex (Gao et al., 2016). Previous studies have shown that the agonist-independent activity of mGluR1 plays a pivotal role in homeostatic intrinsic plasticity throughI
(Shim et al., 2016). However, the relationship between mGluR1 and HCN1 channel after SE is poorly understood. Our results showed that EAAT2 protein was significantly upregulated at 60 minutes post-SE. Increased extracellular glutamate concentrations activate mGluR1 (Ji et al., 2012). mGluR1 upregulation may reflect a subtle shift in the responsiveness of hippocampal neurons to glutamate (Akbar et al., 1996). The upregulation of mGluR1 was most obvious at 1 day post-SE. Moreover, our results revealed that mGluR1 was co-localized with HCN1 in hippocampal CA1 neurons and that this upregulation after SE was accompanied by the downregulation of HCN1 channel. HCN1 exhibited a contrasting pattern of expression to mGluR1 post-SE, indicating a possible correlation. Experiments with a mGluR1 agonist and antagonist revealed that the activation of mGluR1 downregulated HCN1 and increased excitability in PTZ-induced SE. These results were consistent with those obtained in an SE model, suggesting that increased glutamate post-SE activated the expression of mGluR1 and increased the excitability of neurons by downregulating the expression of HCN1 channel. A previous study reported that a mGluR1 antagonist was a potential anticonvulsant agent (Chapman et al., 1999), which inhibited and restored mGluR1 and HCN1 expression, respectively, and suppressed excitability in PTZ-induced SE.We asked how the activation of mGluR1 leads to the downregulation of HCN1 channel in the CA1 region of the hippocampus in PTZ-induced SE. There may be two potential pathways. The first may involve downregulating the transcription of HCN1 mRNA. A previous study showed that HCN mRNA decreased in a subtype- and region-specific manner at all time points after KAinduced SE (Powell et al., 2008). Second, the activation of mGluR1 increases protein kinase activity, leading to the internalization of HCN1 protein. Previous studies have demonstrated that a variety of serine/threonine kinases are activated by mGluRs, including PKA (Li et al., 2011), PKC (Gao et al., 2016), and mitogen-activated protein kinases (Aramori and Nakanishi, 1992; Peavy and Conn, 1998; Dale et al., 2000). Elevated PKC activity is the most canonical example of protein kinase activation by mGluRs (Gao et al., 2016; Li et al., 2017). Apart from PKC activity, some cases in which group I mGluRs modulated cAMP levels to regulate PKA activity have been documented (Joly et al., 1995; Tateyama and Kubo, 2006; Shim et al., 2016). Additional evidence has shown that the coupling of mGluR1 to Gprotein activates adenylyl cyclase, causing cAMP accumulation and PKA activation (Aramori and Nakanishi, 1992; Tateyama and Kubo, 2006; Sugiyama et al., 2008). Upstream regulators of HCN channels include PKA (Narayanan et al., 2010), TRIP8b (Santoro et al., 2009), and Ca/calmodulin-dependent protein kinase (Fan et al., 2005). The results of the present study revealed an increase in the cAMP concentration after SE, which was accompanied by elevated PKA, especially in the DHPG + PTZ group. Therefore, we hypothesized that the upregulation and stimulation of mGluR1 over-activated cAMP-PKA, thereby inhibiting HCN1 channel and either inducing or promoting the hyperexcitability of CA1 neurons after SE. Using a mGluR1 agonist and antagonist, we confirmed that enhancing mGluR1 activity inhibited HCN1 channel and enhanced neuronal excitability. This effect was blocked by a PKA inhibitor. H89 successfully reduced the mGluR1 level, downregulated cAMP-PKA expression, and reversed HCN1 channel inhibition, thereby suppressing the severity and prolonging latency of PTZ-induced SE.
The mechanism through which mGluR1 regulates HCN1 in SE may involve changes in intracellular transport. Recent studies have shown that TRIP8b plays an important role in HCN channels transport by interacting with the COOH terminal of HCN channels (Zerial and McBride, 2001; Santoro et al., 2004). TRIP8b subtypes interact with functional combinations of HCN channels to either strongly upregulate or downregulate HCN channels in the cell membrane (Santoro et al., 2009). Specifically, TRIP8b (1a-4) increases HCN1 surface expression (Zolles et al., 2009), whereas TRIP8b (1b-2) inhibits the transport of HCN1 channel from the plasma membrane (Biel et al., 2009), thereby strongly downregulating HCN1 expression on the cell membrane. The present results revealed the downregulation and upregulation of TRIP8b (1a-4) and TRIP8b (1b-2) proteins, respectively, in the hippocampal CA1 region of rats in the NS + PTZ group. The reduction in TRIP8b (1a-4) levels weakened the inhibition of surface HCN1 channel trafficking to the plasma membrane, whereas the upregulation of TRIP8b (1b-2) promoted this phenomenon. A previous study showed that TRIP8b (1b-2) induces the nearcomplete internalization of HCN1 from the plasma membrane (Santoro et al., 2004). Our results revealed that TRIP8b (1b-2) was significantly upregulated after SE in the NS + PTZ group, but HCN1 surface protein did not completely disappear. In the H89 + PTZ group, H89 inhibited PKA, downregulated PKA protein, upregulated TRIP8b (1a-4) expression, and partly restored TRIP8b (1b-2) protein levels. TRIP8b (1b-2) was still highly expressed, but HCN1 surface protein was downregulated. These phenomena might be attributed to the fact that TRIP8b (1a-4) accounts for 30‒40% and TRIP8b (1b-2) accounts for approximately 10‒15% of total TRIP8b mRNAs in the brain (Santoro et al., 2009). Because TRIP8b (1a-4) is the most abundant TRIP8b subunit, it may play an important role in regulating HCN1 surface protein. A previous study revealed that other TRIP8b isoforms also regulate the expression of HCN1 surface protein (Li et al., 2014). Therefore, the trafficking of HCN1 surface protein might be attributed to a combination of all TRIP8b subunits, especially TRIP8b (1a-4).
Although this study did not measure neuroexcitability using an electrophysiological method, we also concluded that the activation of mGluR1 after SE downregulates the surface expression of HCN1 protein through the following mechanisms: SE occurrence elevates glutamate production, upregulates mGluR1 expression, and subsequently activates the cAMP-PKA signaling pathway. Consequently, TRIP8b (1a-4) and TRIP8b (1b-2) proteins are downregulated and upregulated, respectively, thereby reducing the surface expression of HCN1 and increasing neuronal excitability.
Author contributions:
Study conception and design: YW, XDL, TX; administrative support: XDL, TX, MGM; materials provision: XDL, TX; data collection and assembly: XDL, SJL; data analysis and interpretation: YW, MLC; manuscript writing: XDL. All authors contributed to manuscript revision, read and approved the submitted version of the manuscript.
Conflicts of interest:
The authors declare no competing financial interests.
Availability of data and materials:
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:
Mohd Salman, The University of Tennessee Health Science Center, USA.
Additional file:
Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- Notice of Retraction
- Neuroprotective role of Noggin in spinal cord injury
- Combined cell-based therapy strategies for the treatment of Parkinson’s disease: focus on mesenchymal stromal cells
- Lights at night: does photobiomodulation improve sleep?
- β2-Microglobulin exacerbates neuroinflammation, brain damage, and cognitive impairment after stroke in rats
- The lymphatic drainage systems in the brain: a novel target for ischemic stroke?