
Strategies to treat neurodegeneration in neuronal ceroid lipofuscinosis: a view onto the retina
2022-11-11 05:27UdoBartsch
中国神经再生研究(英文版) 2023年3期
Udo Bartsch
Neuronal ceroid lipofuscinosis (NCL), also known as Batten disease, is the umbrella term for a group of neurodegenerative lysosomal storage disorders with onset mainly in childhood. The total 13 genetically distinct NCLs are caused by mutations in genes encoding soluble or transmembrane proteins, and have been classified according to the affected gene into CLN1 to CLN8 and CLN10 to CLN14. Intracellular accumulation of autofluorescent storage material due to lysosomal dysfunction is a hallmark of all NCLs. Patients affected by this fatal life-limiting disorder typically present with mental retardation, motor impairment, epileptic seizures, and brain atrophy. Progressive retinal degeneration and vision loss are other hallmarks of the majority of NCLs (for details, see Kohlschutter et al., 2019). Rare cases of patients presenting with non-syndromic retinal dystrophies indicate that the retina is particularly sensitive to lysosomal dysfunctions in some NCLs. With the only exception of a brain-directed enzyme replacement therapy (ERT) for CLN2 disease, a condition caused by dysfunctions of the lysosomal enzyme tripeptidyl peptidase 1 (TPP1), there are currently no approved treatment options for these fatal disorders. Studies on animal models of different NCLs have demonstrated the efficacy of various treatment strategies to attenuate neurodegeneration in the brain, and some of them are currently being evaluated in clinical trials (Kohlschutter et al., 2019). The preclinical data and first results from an ERT and a gene therapy trial on CLN2 patients suggest, however, that brain-directed treatments have no or only a minor therapeutic impact on retinal degeneration and vision loss (discussed in Liu et al., 2022). Thus, there is a need to combine retina- and braindirected therapies to combat both vision loss and neurological symptoms. In fact, preservation of vision not only requires preservation of retina function but additionally functional preservation of the visual centers in the brain.
Gene therapy, immunomodulatory therapy, and neuroprotection are among the strategies that have been tested in preclinical studies for their ability to ameliorate retinal dystrophies in NCLs caused by deficiencies or dysfunctions of transmembrane proteins (for a discussion of the latter two approaches, see Kohlschutter et al., 2019). CLN6 is a polytopic transmembrane protein of the endoplasmic reticulum which in a complex with CLN8 recruits lysosomal enzymes for export from the endoplasmic reticulum to the Golgi complex. A naturally occurring mouse model of CLN6 disease, theCln6
mouse, displays an early-onset and rapidly progressing loss of photoreceptors and a subsequent loss of bipolar cells. Unexpectedly, an adenoassociated virus (AAV) 8-mediated expression of CLN6 in photoreceptors had no effect on the progression of the retinal dystrophy. In contrast, photoreceptor degeneration and visual decline as assessed in electroretinogram recordings were significantly slowed when CLN6 was expressed specifically in bipolar cells, indicating that a defect in bipolar cells is the cause of photoreceptor degeneration in CLN6 disease (Kleine Holthaus et al., 2018). Another study showed that intracerebroventricular injections of an AAV9 vector encoding human CLN6 in neonatalCln6
mice attenuated the pathology in visual centers of the brain. Of note, the brain-directed treatment additionally resulted in the expression of CLN6 in multiple retinal cell types and some preservation of photoreceptors. Partial preservation of visual acuity as indicated by improved optokinetic responses of treatedCln6
mice might thus be the result of the combined treatment effects on the brain and retina (White et al., 2021). This study is currently the only example of a beneficial albeit limited therapeutic impact of a braindirected therapy on retinal pathology. Contrary to the partial amelioration of the retinal pathology achieved with the brain-directed AAV9.CLN6 treatment inCln6
mice, AAV9-mediated ocular gene therapy had no effect on photoreceptor survival and retinal function in a CLN6 sheep model (Murray et al., 2021). The most prevalent NCL, CLN3 disease, is caused by mutations in a lysosomal/endosomal transmembrane protein of unknown function. While CLN3 patients usually present with progressive visual deterioration at the early stages of the disease, CLN3 mouse models display a relatively mild retinal dystrophy with compromised inner retinal function and degeneration of bipolar cells. Intravitreal gene therapy targeting CLN3 expression to bipolar cells in a presymptomatic CLN3-deficient mouse model ameliorated inner retinal dysfunction and slowed bipolar cell degeneration (Kleine Holthaus et al., 2020). Together, preclinical data demonstrate the therapeutic potential of AAV-based ocular gene therapy approaches for the treatment of retinal dystrophies in a subgroup of NCLs caused by mutations in genes coding for transmembrane proteins.Enzyme substitution strategies are promising treatment options for NCLs caused by deficiencies or dysfunctions of lysosomal enzymes, as functional lysosomal enzymes administered to diseased tissues are internalized by affected cells through receptor-mediated endocytosis and delivered to lysosomes, eventually resulting in correction of the lysosomal defect. CLN10 disease is caused by mutations in the gene coding for cathepsin D (CTSD), one of the major lysosomal proteases. Complete loss of CTSD enzymatic activity causes congenital NCL with death in early infancy, while partial loss of CTSD enzymatic activity causes CLN10 disease with late infantile, juvenile, or adult onset. Affected patients present with retinal dystrophy that shows similarities to retinitis pigmentosa. TheCtsd
knockout mouse represents an animal model of congenital NCL which faithfully recapitulates key pathologic features of the human disease, including earlyonset accumulation of storage material, reactive astrogliosis, and microgliosis, and brain atrophy. In the retina, loss of cone and rod photoreceptors is followed by the degeneration of retinal interneurons and ganglion cells, resulting in a severely atrophied retina at postnatal day (P) 25, shortly before the animals´ death (Bassal et al., 2021).We have recently compared the efficacy of three enzyme substitution strategies in attenuating the rapidly progressing retinal dystrophy inCtsd
knockout mice: (i) intravitreal injections of recombinant human CTSD (Marques et al., 2020), (ii) intravitreal transplantations of neural stem cells (NSCs) modified to stably overexpress murine CTSD and (iii) an AAV vector-mediated gene transfer of murine CTSD to retinal glial cells and retinal pigment epithelial cells (Liu et al., 2022). To evaluate the potential of an ERT to slow the rapidly progressing retinal dystrophy, recombinant human CTSD was intravitreally injected into the mutant mice at P7 and P14. Analyses of treated eyes at P23 revealed reduced levels of storage material, partial correction of the lysosomal hypertrophy, clearance of accumulated autophagic substrates as indicated by reduced levels of the selective autophagy receptor sequestosome 1/p62 (SQSTM1/p62), and attenuation of neuroinflammation. However, despite the partial correction of various pathological markers, the ERT approach had no impact on the progression of retinal degeneration (Marques et al., 2020). In striking difference, repeated intravitreal injections of recombinant human TPP1 at intervals of three or more weeks resulted in the preservation of retinal structure and function in a CLN2 dog model (Whiting et al., 2020b). Interestingly, intravitreal ERT was also effective in the CLN2 canine model when the treatment was started after the onset of the retinal dystrophy, with a single injection of recombinant human TPP1 attenuating the progression of the retinal pathology over 4 months (Whiting et al., 2020a). The strikingly different treatment efficacy in the CLN10 mouse model and the CLN2 dog model might be related to the more severe and extremely rapidly progressing retinal pathology, the pronounced brain pathology and/or the severe peripheral pathology with lymphopenia and atrophy of visceral organs in the former model. Alternatively, results might indicate that markedly different levels of enzymatic activity need to be restored in different NCLs to achieve significant therapeutic benefits. For CLN2 disease, analyses of genotype-phenotype relationships suggest that restoration of TPP1 enzymatic activity to 10% or less of normal is sufficient to attenuate disease progression. We, therefore, argued that a continuous intravitreal administration of CTSD might be more effective in attenuating retinal degeneration in theCtsd
knockout mutant. To this aim, NSCs were lentivirally modified to overexpress CTSD and transplanted into the vitreous cavity at P7. While the cell-based treatment reduced lysosomal storage accumulation and attenuated lysosomal hypertrophy and neuroinflammation, it neither restored the disrupted autophagic flux nor preserved retina structure albeit CTSD enzymatic activity accounted for ~44% of normal at the time of analysis at P22 (Liu et al., 2022). Possible explanations for this paradoxical observation include (i) preferential uptake of the protease by cell types facing the vitreous cavity (i.e. astrocytes, Müller cells, ganglion cells, and displaced amacrine cells) and/or (ii) slow diffusion of functional CTSD from the vitreous into the retina, resulting in significant enzymatic activity levels only at late stages of the retinal dystrophy (Liu et al., 2022). Different from the limited therapeutic outcome of the cell-based treatment inCtsd
knockout mice, a single intravitreal injection of TPP1 overexpressing autologous mesenchymal stem cells effectively attenuated the progression of the retinal dystrophy for more than 6 months in the CLN2 canine model (Tracy et al., 2016), indicating thatex vivo
gene therapy is among the potential treatment options for vision loss in certain NCLs.The limited outcome of the ERT and the NSCbased enzyme substitution approach in theCtsd
knockout retina might be due to insufficient amounts of functional CTSD during the early stages of the retinal dystrophy. We tested this possibility with an intravitreal gene therapy approach using a self-complementary AAVshH10 vector encoding CTSD under the control of the strong CMV promoter to target expression of the protease to retinal pigment epithelial cells and Müller cells (Liu et al., 2022). Significant CTSD expression was apparent at P9, 4 days after intravitreal injections of scAAVshH10.CTSD. At P22, the protease was detectable in all retinal cell types demonstrating the release of CTSD from retinal glial cells and retinal pigment epithelial cells and subsequent uptake by neighboring cells, a mechanism referred to as cross correction. Of note, levels of CTSD enzymatic activity were similar to those found in healthy wild-type retinas, accumulation of storage material was completely prevented (Figure 1A–D
), and lysosomal hypertrophy and neuroinflammation were markedly attenuated. Furthermore, and different from the NSC-based enzyme substitution strategy, the intravitreal gene therapy fully restored the disrupted autophagic flux as indicated by normalized levels of the autophagy marker SQSTM1/p62 (Figure 1E
andF
) and microtubule-associated protein 1 light chain 3-II. More importantly, the treatment effectively promoted cone and rod photoreceptor survival (Figure 1G
andH
) and significantly slowed the loss of rod bipolar cells (Liu et al., 2022), demonstrating the potential of ocular gene therapy to ameliorate this severe and rapidly progressing retinal dystrophy. An AAV vectormediated gene transfer of lysosomal enzymes or secreted proteins to dystrophic retinas has also shown promise in animal models of other NCLs. For instance, an AAV2 vector-mediated expression of tripeptidyl peptidase 1 (PPT1) ‒ the enzyme affected in CLN1 disease ‒ in retinal ganglion cells ofPpt1
knockout mice slowed the decline in retinal function as demonstrated by electroretinogram recordings and resulted in a better organization of the photoreceptor layer (Griffey et al., 2005). Ocular gene therapy also attenuated deterioration of retinal structure and function in sheep deficient in CLN5, a putative lysosomal enzyme. Intravitreal injections of scAAV9.CLN5 in presymptomatic animals prevented lysosomal storage accumulation, ameliorated reactive astrogliosis, markedly preserved visual function as evaluated in electroretinogram recordings and significantly slowed photoreceptor degeneration (Murray et al., 2021). CLN11 disease is caused by biallelic loss-of-function mutations in theGRN
gene encoding progranulin (PGRN), a secreted protein implicated in the regulation of cell survival, inflammation, and lysosomal homeostasis. While an intravenous gene therapy in young postnatalGrn
knockout mice using an AAV9-2YF capsid attenuated retinal thinning, retina structure was either not preserved or even adversely affected after intravitreal injections of AAV2-7m8.PGRN in mutant mice aged one month or older. Findings indicate that the therapeutic outcome in this CLN11 animal model is determined by the route of vector administration and/or age at treatment.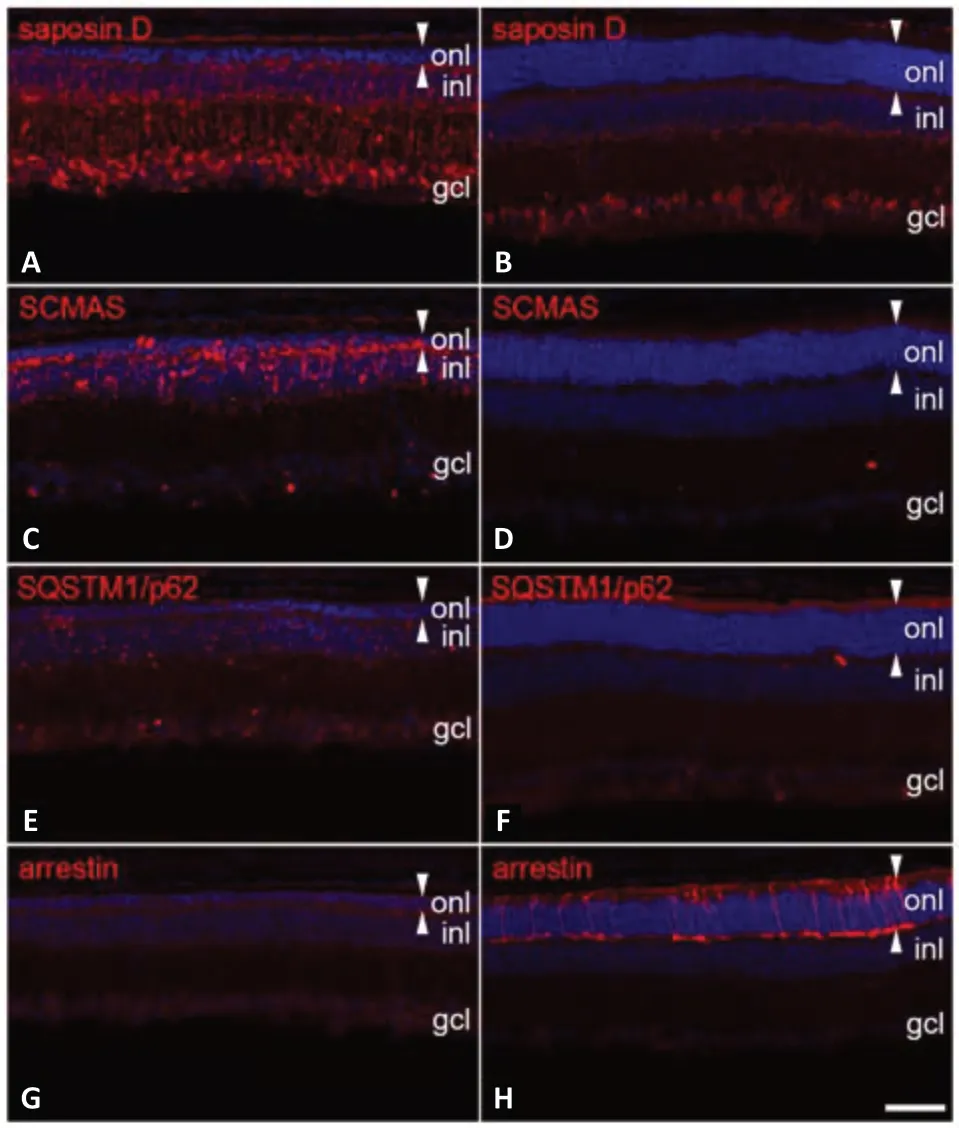
Figure 1 |Ocular gene therapy restores the autophagy-lysosomal pathway and slows photoreceptor loss in Ctsd knockout mice.A scAAVshH10 vector encoding CTSD was intravitreally injected at postnatal day 5, and retinas were analyzed at postnatal day 22 (B, D, F, H). Injections of a scAAVshH10 vector encoding the green fluorescent protein (GFP) into the contralateral eyes served as a control (A, C, E, G). Normalized levels of saposin D (B) and subunit of mitochondrial ATP synthase (SCMAS; D), and the absence of sequestosome1/p62 (SQSTM1/p62) bodies (F) in treated retinas demonstrate prevention of storage material accumulation and clearance of autophagic substrates, respectively. Restoration of the autophagy-lysosomal pathway resulted in significant preservation of the photoreceptor cell layer (onl; outlined with white arrowheads) and survival of numerous arrestin-positive cones (H). gcl: Ganglion cell layer; inl: inner nuclear layer. Scale bar: 50 µm. Unpublished data.
Preclinical studies have demonstrated the efficacy of various therapeutic strategies in attenuating retinal degeneration and vision loss in NCLs caused by dysfunctions of transmembrane or soluble proteins. Whether these strategies will show similar therapeutic benefits in NCL patients remains to be seen in future clinical trials. If successful, meaningful preservation of retina structure and function would improve the quality of life of NCL patients presenting with nonsyndromic retinal dystrophies or with vision loss at thr early stages of the disease. Evidently, effective retina-directed treatment options will be of great relevance once therapies for neurodegeneration in the brain become available, as exemplified by the brain-directed ERT for CLN2 disease.
The author is grateful to all colleagues in the lab for helpful discussions.
This work was supported by the “Stiftung zur
Förderung der Universitätsmedizin Hamburg” and the “Ernst and Berta Grimmke Stiftung” (to UB).
Udo Bartsch
Department of Ophthalmology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
Correspondence to:
Udo Bartsch, PhD, ubartsch@uke.de.https://orcid.org/0000-0003-3227-7568 (Udo Bartsch)
Date of submission:
April 5, 2022Date of decision:
May 25, 2022Date of acceptance:
June 13, 2022Date of web publication:
August 2, 2022https://doi.org/10.4103/1673-5374.350202
How to cite this article:
Bartsch U (2023) Strategies to treat neurodegeneration in neuronal ceroid lipofuscinosis: a view onto the retina. Neural Regen Res 18(3):558-559.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Notice of Retraction
- Neuroprotective role of Noggin in spinal cord injury
- Combined cell-based therapy strategies for the treatment of Parkinson’s disease: focus on mesenchymal stromal cells
- Lights at night: does photobiomodulation improve sleep?
- β2-Microglobulin exacerbates neuroinflammation, brain damage, and cognitive impairment after stroke in rats
- The lymphatic drainage systems in the brain: a novel target for ischemic stroke?