
Axon degeneration: new actor in an old play
2022-11-11 05:27MarinaHerwerthMatthiasWyss
中国神经再生研究(英文版) 2023年3期
Marina Herwerth, Matthias T. Wyss
After an insult of white matter tracts, e.g. in the spinal cord or optic nerve, axons react in general by the activation of a tightly regulated self-destruction program. This so-called axon degeneration cascade can be triggered by various causes, including injury, toxins, and genetic defects, and is a shared pathway in many different neurological diseases (Coleman and Hoke, 2020). Axonal degeneration is thought to be responsible for disease progression and accumulation of disability across many neurological conditions. The hallmark of early axonal injury is the appearance of local spheroid formations along the axon, often referred to as a “pearl-on-string” pattern or axonal beading or swelling. Although this striking shape change has been observed after various types of injury, such as mechanical, chemical, or inflammatory stimuli, we know little about its exact mechanism and its immediate impact on axonal functionality. In this perspective, we would like to contrast the classical calcium-dependent form of axonal degeneration with a recently described form of a calcium-independent mechanism underlying axonal beading.
Calcium-dependent axonal beading:
The key trigger of axonal beading is thought to be an increase in calcium concentration in the acute phase of axonal injury (Kerschensteiner et al., 2005). Calcium enters the axon either from external sources through mechanopores or nanoruptures (Williams et al., 2014), calciumpermeable receptors, or from internal stores such as axoplasmic reticulum or mitochondria (Wang et al., 2012). In various neurological conditions, including trauma, ischemia, and multiple sclerosis, different sources eventually lead to the early elevation of axonal calcium (Figure 1A
). The consecutive downstream cascade is well understood and includes activation of calcium-dependent proteases (calpain proteases) that finally lead to a cytoskeletal breakdown, accumulation of organelles within axonal swellings and eventually axonal loss (Wang et al., 2012). More precisely, cytoskeletal disorganization can include calcium-dependent neurofilament compaction, microtubular disassembly, and actin remodeling, depending on the specific context.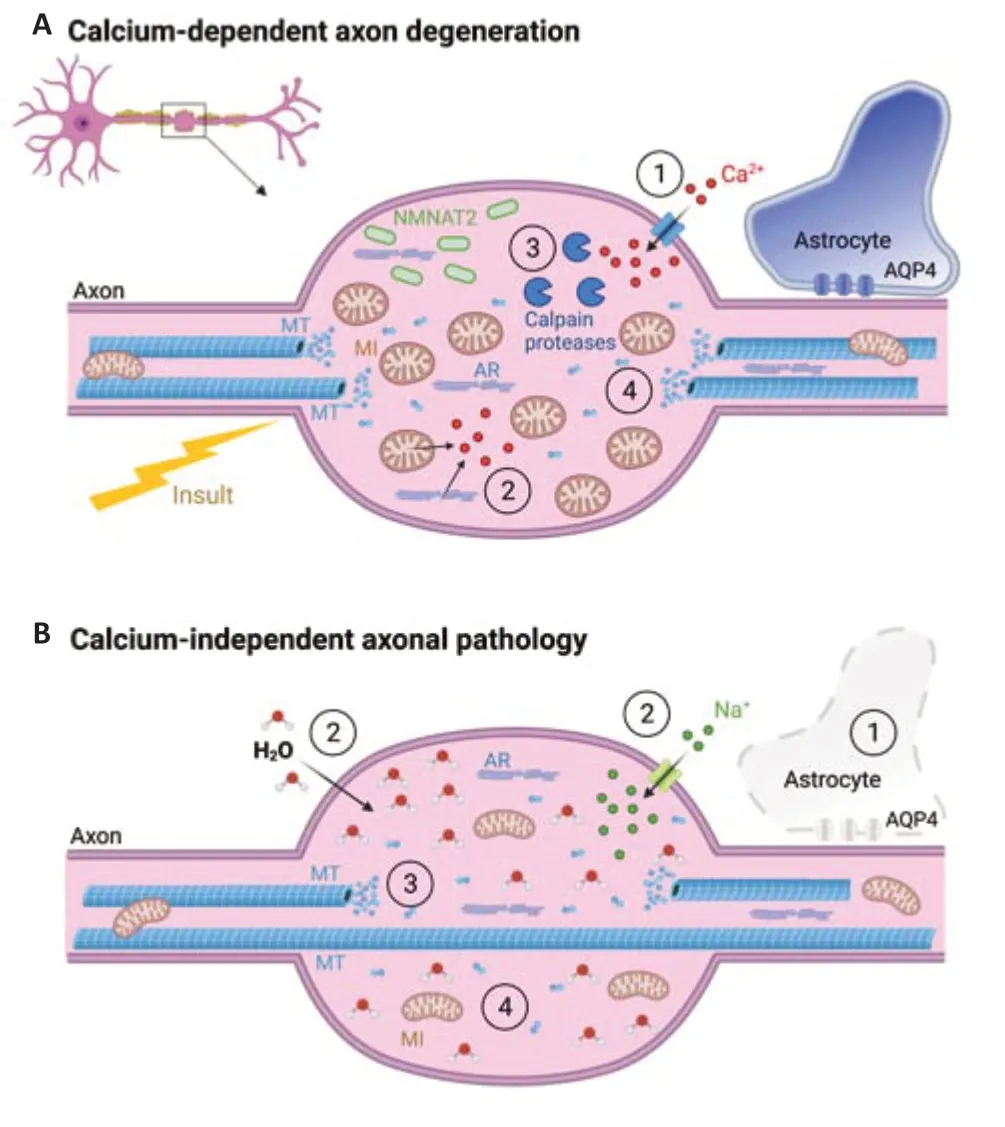
Figure 1 |Early phase of axon degeneration can appear in different forms. (A) Established programs of axon degeneration (such as Wallerian degeneration or focal acute axonal degeneration) are calcium-dependent and are finally characterized by the following sequence of events: Initial injury trigger leads to an abnormal calcium influx through mechanochannels/nanopores (1) and calcium release from internal calcium stores, such as mitochondria and axoplasmic reticulum (2). The increase in intra-axonal calcium level activates calciumdependent calpain proteases (3) that lead to cytoskeletal breakdown and eventually cargo accumulation (4), such as mitochondria and NMNAT2. (B) Astrocytopathy-driven axonal beading has characteristics that distinguish it from the other described pathways. After the lytic depletion of astrocytes (1), a sodium influx-related osmotic challenge (2) induces remodeling of the microtubular cytoskeleton (3) in the initial state without overt cargo accumulation (4). The described processes affect both myelinated and non-myelinated axons. However, for better readability of the figure, the myelin layer has been omitted in the inset illustrations. AQP4: Aquaporin 4 channel; AR: axoplasmic reticulum; MI: mitochondria; MT: microtubules; NMNAT2: the protein nicotinamide mononucleotide adenylyltransferase 2. Created with BioRender.com.
The classical form of such an axonal destruction mechanism is the Wallerian degeneration, in which the segment of an axon dies which is located distally to the site of transection. The proximal neuronal soma survives and can sprout out if the myelin layer is still intact (Coleman and Hoke, 2020). We know that Wallerian-like degeneration does not necessarily require a transection but is rather due to an impairment of the delivery of cargoes for various reasons. Indeed, a blockade of axonal transport is observed in traumatic spinal cord injury (Kerschensteiner et al., 2005), here socalled acute axonal degeneration, in experimental autoimmune encephalomyelitis (Nikic et al., 2011), here so-called focal acute axonal degeneration or neurodegenerative diseases such as amyotrophic lateral sclerosis. Hereby, especially the transport deficit of the protein nicotinamide mononucleotide adenylyltransferase 2 has ultimate consequences, as it is an essential protein for axonal survival. In healthy axons, this protein inhibits the activation of the axonal protein SARM1 (sterile-α and Toll/interleukin 1 receptor motif containing protein 1), which disinhibition launches killing machinery within the axon (Osterloh et al., 2012).
However, this cascade seems not to be a oneway road as one might think. Recently, twophoton microscopy in mice has demonstrated in experimental autoimmune encephalomyelitis (Nikic et al., 2011) and traumatic spinal cord injury (Williams et al., 2014), that after an initial calcium rise, a substantial proportion of axonal swellings can spontaneously recover. Therefore, the phenomenon of axonal beading may represent a metastable state of axonal pathology - at least in the early phase of axonal injury - and thus might be reversible, raising the question of a potential therapeutic window.
Axonal beading without calcium rise?
In all forms of axonal degeneration mentioned so far, an initial intra-axonal calcium accumulation plays a critical role in axonal swelling formation. Intriguingly, studying early axonal pathology in the autoimmune neuromyelitis optica spectrum disorder (NMOSD), Herwerth et al. (2022) found a new form of axonal pathology that makes us rethink this paradigm. NMOSD is a neuroinflammatory disease, in which patients suffer from severe relapses with poor recovery, resulting in accumulating neurological deficits that are likely due to neuronal damage. Interestingly, most NMOSD patients have serum autoantibodies that are directed against the water channel aquaporin-4 (AQP4-Ab; Lennon et al., 2004). As AQP4 is an astrocyte-specific protein in the central nervous system, NMOSD at first glance represents a clear astrocytopathy disease not necessarily involving neurons. It is known that upon binding, AQP4-Ab lead to complementmediated astrocyte necrosis and subsequent inflammation ‒ a hallmark of human NMOSD lesion histopathology (Carnero Contentti and Correale, 2021). Finally, NMOSD lesions show signs of demyelination and a decrease in axon density. However, it remains unclear how the pathology in NMOSD patients spreads from initial astrocytopathy to neuronal damage.To investigate this question, the authors used spinalin vivo
two-photon imaging of astrocytopathic lesions in a recently established acute spinal cord mouse model of NMOSD (Herwerth et al., 2016). After AQP4-Ab-mediated astrocyte loss, axons developed a “pearls-onstring” pattern, which was observed especially in small caliber axons independently of myelination. Surprisingly, functional imaging revealed that calcium homeostasis in most beaded axons was preserved during lesion formation (Herwerth et al., 2022). In the small fraction of axons showing elevations in calcium level, the increases occurred with substantial delay after swelling formation. In this regard, this calcium-independent form of axonal pathology is distinct from known mechanisms of calcium-driven swelling formation.What is the basis for the calcium-independent axonal beading?
Looking directly at the content in such axonal swellings via transmission electron microscopy, the authors could detect notable ultrastructural features of the axonal beading. In contrast to axonal swellings due to collapsed transport of cargoes, which lead to accumulation of different organelles, as observed in multiple sclerosis or trauma, here only a few organelles could be found inside the edematous swellings (Figure 1B
). Using different pharmacological and genetic tools, Herwerth et al. (2022) showed that the mechanisms underlying this form of axonal swelling formation include an ionoosmotic overload leading to a local cytoskeletal remodeling. This pathway is obviously neither shared by the Wallerian degeneration machinery, as SARM1 knockout failed to affect axonal beading formation in NMOSD, nor by non-Wallerian caspase-dependent pathways (Herwerth et al., 2022). This cytoplasmic expansion likely originates from cellular edema due to neuroinflammation in the absence of astrocytic AQP4 channels that play a crucial role in water homeostasis. Besides that, astrocytes are known to have many different metabolic functions in close interaction with neurons. They provide continuous metabolic supply, clear the extracellular space from released glutamate and metabolic waste, control ion/water homeostasis and produce trophic factors, transmitters and transporters that modulate neuronal activity (Verkhratsky and Nedergaard, 2018). Thus, it is not surprising that their absence could influence axon integrity. In this respect, this astrocytopathy-driven axon injury pathway is new and distinct from those previously described in neuroinflammation, neurodegeneration, or trauma, demonstrating that axonal beading can also be induced by astrocytic lysis in a neuroinflammatory environment.Regeneration?
A better understanding of the respective pathways involved in axon degeneration ‒ especially in the early phase of axonal injury ‒ inspired different groups to develop neuroprotective strategies to prevent or reverse axonal damage. Pharmacologically, calcium channel blockers and inhibitors of calcium-dependent calpain proteases have been used effectively to reduce axonal beading early in lesion development after spinal cord trauma (Kerschensteiner et al., 2005; Williams et al., 2014). Knocking out the key inducer of Wallerian degeneration SARM1 (Osterloh et al., 2012) or its pharmacological inhibition (Hughes et al., 2021) has demonstrated a remarkable reduction in axonal degeneration as well. The neutralization of reactive oxygen species molecules (Nikic et al., 2011) during neuroinflammation in experimental autoimmune encephalomyelitis is a further promising neuroprotective strategy. In the calcium-independent NMOSD swelling formation, hyperosmolar treatment and sodium channel modulation have demonstrated reliable efficacy in the reduction of axonal beading (Herwerth et al., 2022). Aiming at cytoskeletal remodeling, pharmacological microtubule stabilization successfully diminished the percentage of swollen axons. However, all these therapeutics are limited by their application prior to the injury, their side effects, non-specificity, or toxicity and rather help to understand the underlying mechanism than provide a solid treatment option for axonal recovery. Therefore, they are unlikely to be useful in a clinical context in their actual form. Needless to say, the need for future complementary human studies is substantial, as most of the recent advances in understanding axon degeneration mechanisms originate from animal models.Future perspectives:
The development of new techniques and animal models of different neurological conditions in recent years has remarkably extended our knowledge about axonal degeneration mechanisms. These new findings encourage us to consider axon degeneration as a flexible context-specific program with diverse scenarios rather than a common destruction trunk. Indeed, accumulating evidence suggests that axon degeneration in central nervous system disorders is not as homogeneous and unidirectional as one might assume, but shows heterogeneity in different steps after initial axonal injury: axonal beading can appear after transsection, but also as a reaction to a neuroinflammatory context without transsection at all; it can either rely on a rise in free intra-axonal calcium or also be calciumindependent; and last but not least, in some occasions, it can harbor a potential of recovery within a transient time window.The new actor “astrocyte” on the scene may further lift the curtain of our understanding of axon degeneration and may motivate research for novel neuroprotective treatment strategies. Recognizing astrocytes as an additional factor in neurodegeneration may help to envision new regeneration endeavors. For instance, modulation of astrocyte reactive oxygen species scavenger capacity to decrease oxidative stress or enhancement of excitatory amino acid transport (EAAT1/EAAT2) efficacy to reduce glutamate excitotoxicity may both have the potential to improve neuroregenerative outcomes. Lastly, astrocytes have been shown to develop neuroprotective and neurotoxic properties depending on their interaction with microglia. Modulating microglial signaling therefore may be an additional approach to shape astrocyte performance. Although attempts to develop treatments for neurodegenerative disorders have not yet been clinically successful, the variety of possible targets warrants further translational intervention studies and hopefully will bring one day effective neuroprotective therapy for various neurological conditions.
We would like to apologize to authors whose work could not be included due to space limitation.
Marina Herwerth, Matthias T. Wyss
Institute of Pharmacology and Toxicology, University of Zurich; Neuroscience Center Zurich, University and ETH Zurich, Zürich, Switzerland (Herwerth M, Wyss MT)
Former address: Institute of Neuronal Cell Biology; Department of Neurology, Technische Universität München, Munich, Germany (Herwerth M)
Correspondence to:
Marina Herwerth, MD, marina.herwerth@pharma.uzh.ch.https://orcid.org/0000-0001-5125-4594 (Marina Herwerth)
Date of submission:
April 6, 2022Date of decision:
May 25, 2022Date of acceptance:
June 8, 2022Date of web publication:
August 2, 2022https://doi.org/10.4103/1673-5374.350200
How to cite this article:
Herwerth M, Wyss MT (2023) Axon degeneration: new actor in an old play. Neural Regen Res 18(3):547-548.
Availability of data and materials:
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:
Suzanne Scarlata, Worcester Polytechnic Institute, USA; Senmiao Sun, Harvard Medical School, USA.
Additional files:
Open peer review reports 1 and 2.

- 中国神经再生研究(英文版)的其它文章
- Notice of Retraction
- Neuroprotective role of Noggin in spinal cord injury
- Combined cell-based therapy strategies for the treatment of Parkinson’s disease: focus on mesenchymal stromal cells
- Lights at night: does photobiomodulation improve sleep?
- β2-Microglobulin exacerbates neuroinflammation, brain damage, and cognitive impairment after stroke in rats
- The lymphatic drainage systems in the brain: a novel target for ischemic stroke?