
Technological advances expand our knowledge of lysosomal dysfunction in neurodegeneration
2022-11-11 05:27ChaseChenYuChenEllenSidransky
中国神经再生研究(英文版) 2023年3期
Chase Chen, Yu Chen, Ellen Sidransky
In the almost seven decades since the initial discovery of the lysosome as an organelle, our understanding of the role of lysosomes has greatly evolved. We now know lysosomal function encompasses far more than its traditionally described role as the cell’s “garbage disposal”, referring to its well-established catabolic function. Lysosomes are integral to maintaining cellular health and viability, and they act as a major signaling hub within the cell (Ballabio and Bonifacino, 2020). Lysosomes regulate autophagy, a key mechanism for regulating cellular homeostasis. The aberrant regulation of different lysosomal pathways is frequently observed in neurodegenerative diseases. Studies have identified a multitude of lysosomal genes that are now implicated in disorders including Parkinson’s disease (PD), Alzheimer’s disease (AD), and amyotrophic lateral sclerosis (Udayar et al., 2022). For example, many genes associated with lateonset AD, such asAPOE
andPLD3
, are associated with autophagy-lysosomal pathways (Van Acker et al., 2019). In addition, lysosomal dysfunction contributes to the aggregation of alpha-synuclein in PD and the build-up of beta-amyloid and taucontaining neurofibrillary tangles in AD (Mazzulli et al., 2016; Feng et al., 2020). However, the development of therapeutics targeting PD, AD, and related neurodegenerative diseases has been hindered by the fact that we still do not fully understand the cellular and molecular changes contributing to lysosomal dysfunction in neurodegeneration. Thus, an emphasis has been placed on developing new techniques that can elucidate minute alterations in cellular and molecular processes. Over the past few years, both the development of methods for the rapid isolation of intact lysosomes and new CRISPR/Cas9-based genetic screens have furthered our knowledge of many lysosomal functions. Recently, studies with isolated lysosomes have shown the dynamic nature of nutrient exchange between the lysosome and the cytosol (Abu-Remaileh et al., 2017). In parallel, the first CRISPR interference (CRISPRi)-based genetic screens in human induced pluripotent stem cell (iPSC)-derived neurons have yielded novel insights into the relationship between glycosphingolipid accumulation and elevated oxidative stress (Abu-Remaileh et al., 2017; Tian et al., 2021). These findings pave the way for expanding our knowledge on how lysosomal dysfunction relates to neurodegeneration (Figure 1
).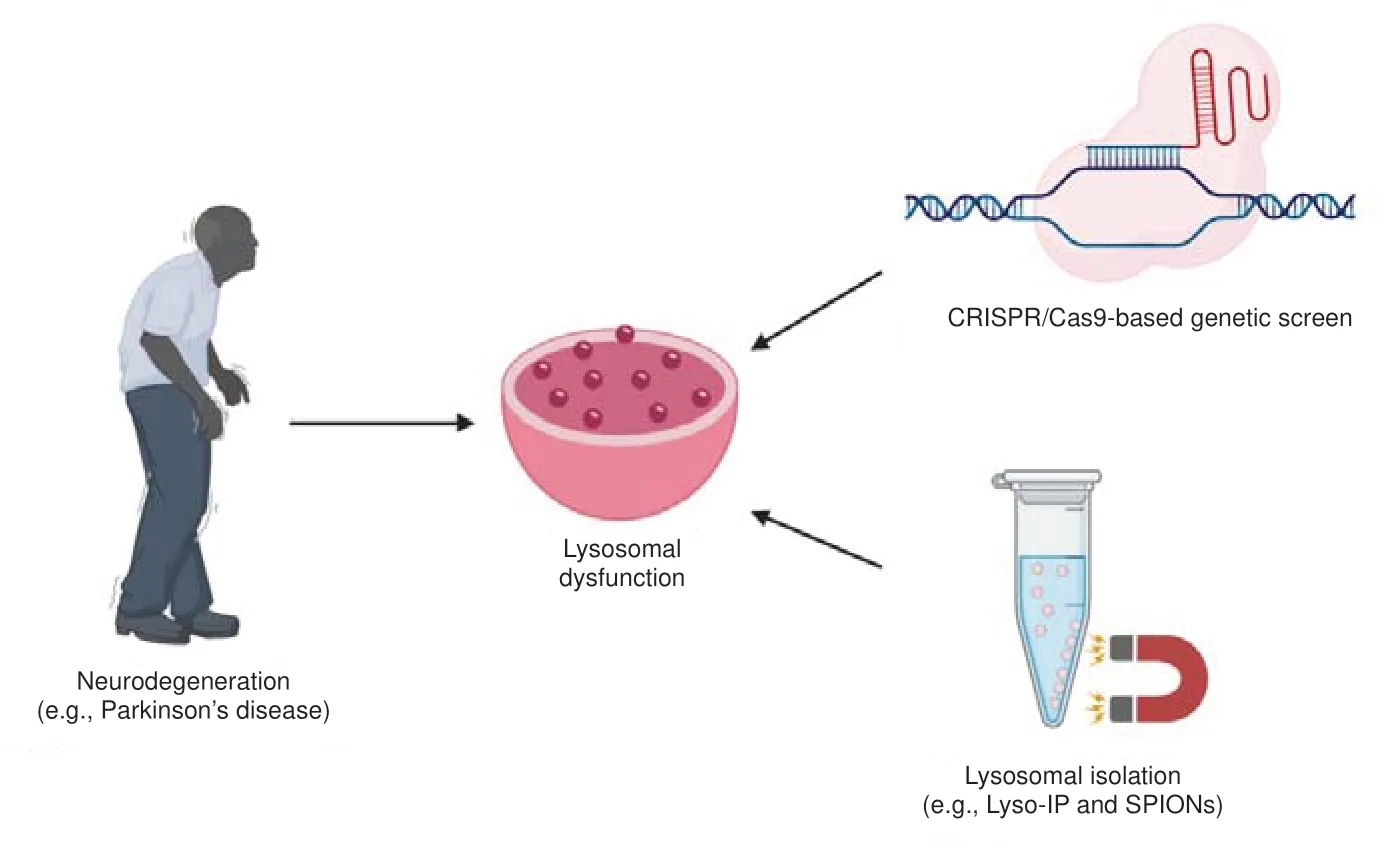
Figure 1 |New means to probe lysosomal dysfunction.Growing evidence has shown that lysosomal dysfunction plays an important role in neurodegeneration. Technological advances in studying lysosomal dysfunction, such as new lysosome purification techniques and CRISPR/Cas9-based genetic screens, may provide new insights into how aberrant lysosome function contributes to neurodegeneration. SPIONs: Superparamagnetic iron oxide nanoparticles.
The recent development of new tools has greatly enhanced our capability to purify whole lysosomes. The capacity to study the lysosome at the organelle level increases our ability to observe minute changes and to detect the presence of low abundance molecules not previously distinguishable in whole-cell samples. The ‘Lyso-IP’ method, first described by Abu-Remaileh et al. (2017), allows for the rapid isolation of intact lysosomes within ten minutes, preserving the labile lysosomal lipidome and metabolome (Chen et al., 2022). Utilizing a lysosomal transmembrane protein fused to a cytosolic 3x-HA epitope tag (TMEM192-3xHA), lysosomes are immunoprecipitated using anti-HA conjugated magnetic beads. Lyso-IP presents a massive leap forward compared to traditional techniques of density-based gradient centrifugation for lysosome isolation, which are both labor-intensive and timeconsuming. In addition, the Lyso-IP method utilizes a liquid chromatography-mass spectrometry compatible ‘KPBS’ buffer, which mimics cytosolic potassium concentrations, stabilizes membrane potential, and preserves labile molecules. Therefore, lysosomes isolated with the Lyso-IP method are optimal for downstream liquid chromatography-mass spectrometry analysis. Many published studies have already successfully used Lyso-IP to prepare lysosomes for proteomic, lipidomic, and metabolomic analyses.
The application of the Lyso-IP method to many diverse cellular models has already provided new insights. Two examples of new perspectives resulting from the use of this technique concern the regulation of the mechanistic target of rapamycin complex 1 (mTORC1) pathway and how cellular regulation is disrupted in the lysosomal storage disorder Niemann Pick C (NPC). The aberrant regulation of the mTORC1 pathway is known to be associated with neurodegeneration, while NPC patients often present with neurological symptoms. The first published study by Abu-Remaileh et al. (2017) using Lyso-IP to isolate lysosomes from human embryonic kidney (HEK-293T) cells showed that the efflux of amino acids and metabolites in the lysosome are controlled via the vacuolar HATPase pump and mTOR-dependent signaling. Furthermore, a separate study also performed with HEK293T cells showed that the lysosomal transmembrane protein SLC38A9 is a critical arginine sensor for the mTORC1 pathway, and Lyso-IP was used to show the central role of SLC38A9 in amino acid homeostasis (Wyant et al., 2017). Other studies looking at NPC-regulation discovered that oxysterol binding protein (OSBP) was responsible for regulating endoplasmic reticulum-lysosome cholesterol trafficking, presenting OSBP as a promising therapeutic target for NPC and related diseases (Lim et al., 2019). In this specific study, Lyso-IP was utilized to show that endogenous OSBP, along with other OSBP-related proteins are associated with the lysosome. These highlighted examples reflect only a few of the recently published works involving Lyso-IP.
A similar tool to isolate mitochondria, known as ‘Mito-IP’, has already been applied to createin vivo
mouse models of disease. The generation of MITO-tag mice has increased our ability to assess mitochondrial metabolismin vivo
and improved our understanding of mitochondrial metabolism (Bayraktar et al., 2019). Similar applications of lysosome isolation in mouse models of neurodegenerative diseases hold immense potential in increasing our knowledge of diseaserelevant mechanisms and are currently under development.A second recently published method to isolate lysosomes involves the nonspecific endocytosis of superparamagnetic iron oxide nanoparticles (SPIONs). SPIONs are nanoparticles that consist of an inorganic magnetic core surrounded by an inorganic or organic shell that have been widely applied for different biomedical applications. Tharkeshwar et al. (2017) discovered that SPIONs coated with dimercaptosuccinic acid could specifically target late endosomes and lysosomes. Then, using dimercaptosuccinic acidcoated SPIONs, the authors purified lysosomes from NPC1-knockout HeLa cells, resulting in the discovery of the accumulation of several species of glycerophospholipids not traditionally associated with NPC1 deficiency.
Another new and powerful tool now being used to study lysosomal dysfunction in neurodegeneration is the use of CRISPR/Cas9-based genetic screens. In short, in CRISPRi-based genetic screens, cells expressing a catalytically dead Cas9 fused with the Krüppel-associated box repressor are transduced with single guide RNAs at a multiplicity of infection such that each cell only receives one single guide RNA, repressing transcription of one gene per cell. Then, paired-ended sequencing is utilized to assess the frequency of perturbations associated with the phenotypes of interest. Recently, the first CRISPRi-based genetic screen in iPSC-derived cortical neurons showed that the knockdown of the lysosomal pre-protein prosaposin led to glycosphingolipid accumulation and elevated oxidative stress within the cell (Tian et al., 2021). More importantly, these phenotypes were found to be unique to cortical neurons and are not seen in other iPSC-derived neuronal cell types. The application of CRISPRi-based genetic screens to other cellular models of disease could prove especially meaningful in understanding lysosomal dysfunction and neurodegeneration.
A prime example for the future application of these tools is in studyingGBA1
-associated PD. Variants in theGBA1
gene, which encodes for the lysosomal hydrolase glucocerebrosidase (GCase), have been shown to be one of the most common genetic risk factors for developing PD (Sidransky et al., 2009). However, the underlying mechanisms linkingGBA1
mutations to the development of PD are still unknown, but two main hypotheses have emerged. The gain-of-function hypothesis suggests that mutated GCase protein directly leads to alphasynuclein accumulation, while the loss-of-function hypothesis suggest mutations to GCase leads to accumulation of its lipid substrate resulting in aggregation of alpha-synuclein. In theory, studying individual isolated lysosomes may help to clarify this mystery by providing the ability to detect subtle changes in lipid and protein concentrations, as well as enabling the identification of low abundance molecules. Furthermore, CRISPR/Cas9-based genetic screens can lead to the identification of new risk loci associated withGBA1
-associated PD that ultimately may provide new mechanistic insights. Many aspects of the link between lysosomal dysfunction and neurodegeneration remain to be elucidated. However, the development of new tools has significantly helped improve our grasp of the cellular and molecular changes underlying neurodegeneration. The different findings briefly highlighted here are surely only the tip of the iceberg. These new tools are likely to still yield deeper insights into disease mechanisms. Ongoing studies applying lysosome isolation and CRISPR/Cas9-based screens to patient-derived iPSCs in disease-relevant cell types are also likely to provide new leads for the development of therapeutics for neurodegenerative disorders.Chase Chen, Yu Chen, Ellen Sidransky
Medical Genetics Branch, National Institutes of Health, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, USA
Correspondence to:
Ellen Sidransky, MD, sidranse@mail.nih.gov.https://orcid.org/0000-0002-3019-8500 (Ellen Sidransky)
Date of submission:
March 18, 2022Date of decision:
April 29, 2022Date of acceptance:
May 16, 2022Date of web publication:
June 2, 2022https://doi.org/10.4103/1673-5374.346490
How to cite this article:
Chen C, Chen Y, Sidransky E (2023) Technological advances expand our knowledge of lysosomal dysfunction in neurodegeneration. Neural Regen Res 18(3):539-540.
Availability of data and materials:
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:
Roberta Ghidoni, IRCCS Centro San Giovanni di Dio Fatebenefratelli, Italy.
Additional file:
Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- Notice of Retraction
- Neuroprotective role of Noggin in spinal cord injury
- Combined cell-based therapy strategies for the treatment of Parkinson’s disease: focus on mesenchymal stromal cells
- Lights at night: does photobiomodulation improve sleep?
- β2-Microglobulin exacerbates neuroinflammation, brain damage, and cognitive impairment after stroke in rats
- The lymphatic drainage systems in the brain: a novel target for ischemic stroke?