
Ferroptosis: a critical player and potential therapeutic target in traumatic brain injury and spinal cord injury
2022-11-11 05:27QingShengLiYanJieJia
中国神经再生研究(英文版) 2023年3期
Qing-Sheng Li , Yan-Jie Jia
Abstract Ferroptosis, a new non-necrotizing programmed cell death (PCD), is driven by iron-dependent phospholipid peroxidation. Ferroptosis plays a key role in secondary traumatic brain injury and secondary spinal cord injury and is closely related to inflammation, immunity, and chronic injuries. The inhibitors against ferroptosis effectively improve iron homeostasis, lipid metabolism, redox stabilization, neuronal remodeling, and functional recovery after trauma. In this review, we elaborate on the latest molecular mechanisms of ferroptosis, emphasize its role in secondary central nervous trauma, and update the medicines used to suppress ferroptosis following injuries.
Key Words: ferroptosis; immune response; inflammation; iron homeostasis; lipid metabolism; medicine; programmed cell death; spinal cord injury; traumatic brain injury

Introduction 506 Retrieval Strategy 506 Mechanisms for Ferroptosis 506 Ferroptosis Plays a Crucial Role following Central Nervous System Trauma 508 Treatments for Ferroptosis following Central Nervous System Trauma 509 Conclusion 510
Introduction
Traumatic brain injury (TBI) and spinal cord injury (SCI) are the primary causes of traumatic injuries to the central nervous system (CNS). From 1990 to 2016, the total global incidence of TBI and SCI increased significantly because of increased falls and road-related injuries (GBD 2016 Traumatic Brain Injury and Spinal Cord Injury Collaborators, 2019). Moreover, the increased incidence of TBI and SCI seems to continue over time, given the growing and aging population. Unfortunately, SCI is currently incurable. Patients with TBI and SCI require a prolonged and high level of specialized nursing. Treatments only maximize residual function by rehabilitating and minimizing secondary complications (Ahuja et al., 2017; Ismael et al., 2021). Similarly, despite promising laboratory results, many multicenter, clinical, randomized controlled trials of medical and surgical interventions for TBI did not have beneficial effects (Maas et al., 2017). Therefore, the long-term burdens of CNS trauma for patients, caregivers, healthcare systems, and the economy are a considerable part of the global diseases burden (Badhiwala et al., 2019).
The primary SCI and the direct physical injury to neurons and axons results in a secondary injury cascade characterized by edema and inflammation that contributes to progressive neuron and glial cell death that spreads beyond the initial impact site. Cystic cavities form a chronic, physical, and chemical barrier surrounding the glial/fibrotic scar and remodel the lesion, to potently stabilize the spread of inflammation and lesion volume, but inhibit axonal regeneration (Tran et al., 2018). The secondary injury also plays a marked role in TBI. For example, neuroinflammation and swelling can cause an increase in intracranial pressure, which blocks blood flow or damages brain tissues. The procedures for secondary injury include glutamate excitotoxicity, calcium overload, mitochondrial dysfunction, and free radical-induced oxidative damage. These are closely related to chronic traumatic encephalopathy (CTE) (Simon et al., 2017) and ferroptosis (Gleitze et al., 2021; Li et al., 2021; Zhang et al., 2021c).
Programmed cell death (PCD) includes apoptosis, necroptosis, autophagy, ferroptosis, pyroptosis, and paraptosis and is known to play a crucial role in eliminating unnecessary and damaged cells in the progress of neurogenesis and neurodegeneration after CNS trauma (Green, 2019; Zhu et al., 2021b). Ferroptosis, a unique iron-dependent form of non-apoptotic PCD, was discovered and named by Dixon et al. (2012). It genetically, biochemically, and morphologically differs from other PCDs. Recent evidence has suggested that ferroptosis is crucial for secondary injuries following both SCI and TBI, but currently, there are few studies. Researching the mechanisms of ferroptosis can enhance understanding of the pathophysiological mechanisms of SCI and TBI and help guide strategies to improve neuron regeneration and recovery of function at acute and chronic phases. This review describes the most recent understanding of ferroptosis and its crucial role following SCI and TBI. Last, we list the latest inhibitors of ferroptosis used in CNS traumatic injuries.
Retrieval Strategy
We searched the literature of articles with the keyword “ferroptosis” published from January 2012 to February 2022 on PubMed. The search terms “spinal cord injuries”, “spinal cord injury”, “spinal cord contusion”, “spinal cord transection”, “spinal cord trauma”, “traumatic brain injuries”, “brain trauma”, and “traumatic encephalopathy” were used for article selection. The results were further screened by the title and abstract, mainly focusing on the studies related to SCIs and TBIs.
Mechanisms for Ferroptosis
Phospholipid peroxidation
Unrestrained accumulation of phospholipid hyperoxides (PLOOHs) is currently the most downstream step of ferroptosis. It may damage membrane integrity, other macromolecules, and cellular structures and cause inflammation or immune response (Jiang et al., 2021). Plasma membrane damage in ferroptosis is related to the formation of membrane nanopores. During this process, an accumulation of cellular Cainduces activation of the endosomal sorting complexes required for transport-III-dependent membrane repair machinery (Pedrera et al., 2021). Therefore, Cafluxes before plasma membrane rupture may be a general feature of PCD, and local endosomal sorting complexes are likely required for transport-III activation as a universal protective mechanism.
Doll et al. (2017) found acyl-coenzyme A (CoA) synthetase long-chain family member 4 (ACSL4) as an essential propellant of ferroptosis. ACSL4 preferentially acylates long-chain polyunsaturated fatty acids (PUFAs), and lysophosphatidylcholine acyltransferase 3 catalyzes acylated long-chain PUFAs inserted into cellular membrane phospholipids (PL). Inhibition of ACSL4 enables cells’ resistance to ferroptosis. This effect may result from a conversion from easily oxidized PUFA to monounsaturated fatty acid (MUFA) in membrane PL (Doll et al., 2017). Therefore, exogenous MUFAs can potently inhibit ferroptosis, and this reaction requires acyl-CoA synthetase long-chain family member 3 acylating MUFAs (Magtanong et al., 2019). Linoleates with conjugated double bonds insert into cellular lipids via acyl-CoA synthetase long-chain family member 1 and induce ferroptosis in diverse cancer cell types (Beatty et al., 2021).
PL contains PUFA inserted on cellular membranes—the primary targets for free radical attack. Factors induce the initial cellular membrane lipids oxidation, including exogenous physical and chemical reagents, enzymes, or electron transport chains in mitochondria and other cellular metabolic processes. Moreover, a PL radical (PL•) forms from these changes. It needs oxygen to yield a PL peroxyl radical (PLOO•) and form PLOOH and new PL•. PLOOH and cellular ferrous ions are substrates of Fenton-like reaction, generating alkoxyl (PLO•) or PLOO• and rapidly amplifying PLOOHs. PLOO• and PLO• can also oxidate PUFA, a chain reaction propagating lipid-free radical (Conrad and Pratt, 2019). In acidic cancer cells, omega-3 and omega-6 PUFA assemble in lipid droplets and induce ferroptosis when they exceed the capacity of triglyceride storage into lipid droplets. These cytotoxic effects aggravate while the inhibitors of diacylglycerol acyltransferase inhibitors (DGATi) intervene in the formation of lipid droplets (Dierge et al., 2021).
Lipoxygenases (LOXs) are the central players in cell death induced by the knockout of glutathione (GSH) peroxidase type 4 (GPX4) (Seiler et al., 2008). However, Friedmann Angeli et al. (2014) proved that the knockdown of arachidonate 5-lipoxygenase (ALOX) 5 and ALOX15—two crucial LOXs—did not impact cell death. Moreover, less than half of diverse LOXs inhibitors can rescue cells that overexpress LOXs, and the cytoprotective effect is likely from their property as radical-trapping antioxidants, rather than their potency to inhibit LOXs (Zilka et al., 2017; Shah et al., 2018). Thus, some authors suggested that non-enzymatic lipid peroxidation may be the critical driver of ferroptosis, and LOX contributes to the initiation and/or propagation of lipid hydroperoxides. Cytochrome P450 oxidoreductase (POR) and nicotinamide adenine dinucleotide-cytochrome b5 reductase 1 (CYB5R1) further challenge the universal role of LOX. P450 oxidoreductase and CYB5R1 fetch electrons from nicotinamide adenine dinucleotide phosphate (NADPH) to oxygen to generate hydrogen peroxide which reacts with irons and peroxidizes membranal PL containing PUFA. Their gene knockout can suppress lipid peroxidation and ferroptosis, and they compensate for the loss of ALOX15 activity (Zou et al., 2020; Yan et al., 2021). Notably, phosphatidylethanolamine binding protein 1 (PEBP1) binds with LOXs shifting their substrate competence, enabling them to directly react with the PL-containing PUFA inserted into membranes (Wenzel et al., 2017). Furthermore, ferrostatin-1 (Fer-1) does not affect ALOX15 alone. It effectively inhibits PLOOH generations via the ALOX15/PEBP1 complex and can disrupt the catalytically required allosteric motions of the ALOX15/PEBP1 complex (Anthonymuthu et al., 2021). Linking to PEBP1 enables LOXs to oxidize membrane PUFA. LOXs are still the key enzymes to catalyze the peroxidation of PUFA (Figure 1
).Iron metabolism
Ferroptosis is dependent on intracellular iron. First, iron is the necessary reactant of the non-enzymatic Fenton chain reaction, critical for lipid peroxidation (Conrad and Pratt, 2019). Second, LOXs and P450 oxidoreductase require iron for catalyzing lipid oxidation (Yosca et al., 2013; Dufrusine et al., 2019). Last, redox-based metabolism also requires iron atoms to generate cellular reactive oxygen species (ROS) (Dixon and Stockwell, 2014).
The transferrin receptor (TFR) regulates cellular iron uptake through endocytosis of iron-loaded transferrin. The lipogenesis regulator sterol regulatory element-binding transcription factor 2 directly mediates transferrin transcription, reducing cellular iron pools, PL peroxidation, and thereby ferroptosis (Hong et al., 2021). The antibody of TFR can stain ferroptotic cells in multiple-cell culture and tissue sections, which is a scarce reliable marker to selectively stain ferroptosis and characterize the extent of ferroptosis (Feng et al., 2020).
Ferritin is the main iron storage protein (Shi et al., 2008). Ferritin autophagy, a selective autophagic degradation towards ferritin, leads to labile iron overload and subsequent lipid peroxidation, which may lead to ferroptosis. This cell death results in autophagy activation and degradation of ferritin and ferritinophagy cargo receptor nuclear receptor coactivator 4 (NCOA4). Inhibiting ferritinophagy by blocking autophagy or NCOA4 can prohibit iron overload, lipid oxidation, and eventual ferroptosis (Ajoolabady et al., 2021). Fang et al. (2021) reported a new ferroptosis inhibitor 9a, which targets NCOA4 and reduces the intracellular ferrous iron by breaking the NCOA4-ferritin H (Fth) interaction. It can ameliorate ischemia/refusion injury in a rat model (Fang et al., 2021). Zhang et al. (2021b) found that coatomer protein complex subunit zeta 1 knockdown increases NCOA4, intracellular ferrous iron, and ferroptosis. Neurons mainly express Fth, whose conditional knockout makes mice viable and fertile with the altered iron environment and more sensitive to ferroptosis after TBI (Rui et al., 2021). Similarly, the downregulation of Fth makes liver cancer cells more sensitive to ferroptosis (Asperti et al., 2021). Furthermore, Fth-knockdown in myocardial cells reduces the expression of solute carrier family 7 member 11 (SLC7A11), the unit of glutamate-cysteine antiporter (System Xc) (Fang et al., 2020), which may result in a vicious circle by damaging both iron homeostasis and GSH metabolism. Currently, only the Fth of ferritin seems involved in ferroptosis protection.
Ferroprotein (FPN) can prevent erastin-induced ferroptosis via exporting intracellular iron, which can be restrained by the inhibitor and knockdown of FPN (Geng et al., 2018). Additionally, miR-124 can decrease FPN expression and aggravated iron overload in the brain post-cerebral hemorrhage, which may be correlated with neurological function damage in aged patients with cerebral hemorrhage (Bao et al., 2020). Silencing the ubiquitin-specific protease 35, associated with cell proliferation and mitosis, functions as a deubiquitinase to maintain FPN stability. Its silencing promotes ferroptosis, and overexpression inhibits erastin/Ras-selective lethal small molecule 3-triggered ferroptosis in lung cancer cells (Tang et al., 2021). Bao et al. (2021a) found that FPN is downregulated in the encephalon of Alzheimer’s patients and mouse models, and loss of this gene induces amyloidβ aggregation, hippocampal atrophy, and memory impairment by promoting ferroptosis.
Poly(rC) binding protein 1, a cytosolic iron chaperone, binds and distributes iron to recipient proteins in cells. The lack of poly(rC) binding protein 1 in mice hepatocytes induces lipogenesis and oxidative-stress pathways in the absence of iron overload, and dietary antioxidant therapy and iron restriction can prevent this process. Therefore, poly(rC) binding protein 1 is crucial for preventing cellular iron toxicity (Protchenko et al., 2021;Figure 1
).Suppressors of ferroptosis
GPX4 is the central suppressor of ferroptosis (Ingold et al., 2018). In neurons, selenium supplementation can augment GPX4 and other selenoprotein genes inhibiting ferroptosis via coordinated activation of the transcription factors activating enhancer binding protein 2 gamma and specificity protein 1 (Alim et al., 2019). Cystine, imported via System Xc, is used to synthesize GSH and CoA, which can downregulate the ferroptosis. Cysteine is formed from cystine. In non-small cell lung cancer cells, deprivation of cyst(e)ine induces accumulation of γ-glutamyl-peptides, which are produced due to an atypical activity of glutamate-cysteine ligase catalytic subunit. The synthesis of γ-glutamyl-peptides limits ferroptosis by preventing the accumulation of glutamate. Nuclear factor erythroid 2-related factor 2 (NRF2) is a critical regulator of the glutamate-cysteine ligase catalytic subunit (Kang et al., 2021). Cystine uptake is mediated by SLC7A11, a system Xcsubunit, and promotes GSH synthesis and directly affects GPX4 protein synthesis. Mechanistically, the rapamycin complex 1, activated by cyst(e)ine, promotes GPX4 synthesis through the Rag-mechanistic target of rapamycin complex 1-eukaryotic translation initiation factor 4e binding protein 1 signaling pathway, and rapamycin complex 1 pharmacologic inhibitor can decrease levels of GPX4 and increase the sensitivity of cancer cells to ferroptosis (Zhang et al., 2021a). The deletion of SLC7A11 induces tumor-selective ferroptosis and inhibits cancer growth (Badgley et al., 2020); similarly, overexpression of SLC7A11 promotes the expression of GSH and inhibits ferroptosis (Fang et al., 2020).
Sensitivity to GPX4 inhibitors varies greatly across cancer cell lines, indicating that other factors resist ferroptosis (Doll et al., 2019). Through the expression cloning approach and CRISPR-Cas9 screen, ferroptosis suppressor protein 1 (FSP1) has been uncovered as a GPX4-independent ferroptosis resistance pathway involving NADPH, FSP1, and ubiquinone that can complement GPX4 deletion. Myristoylated FSP1 is recruited to the organelle membrane, including perinuclear structures, Golgi apparatus, and the plasma membrane. In the membrane, FSP1 suppresses ferroptosis by either reducing ubiquinone (coenzyme Q, CoQ) to form ubiquinol, which can directly trap lipid peroxyl radicals to halt the Fenton reaction, or by restoring α-tocopherol (vitamin E); both processes use electrons from NADPH (Bersuker et al., 2019; Doll et al., 2019).
Metabolomics analyses recently linked the mitochondrial enzyme dihydroorotate dehydrogenase (DHODH) to ferroptosis. DHODH is a flavindependent enzyme containing iron, which plays an essential role in the de novo synthesis of pyrimidines. It alleviates lipid peroxides accumulated via reducing CoQ, which suppresses ferroptosis coordinating with mitochondrial GPX4 in the inner membrane of the mitochondria. Therefore, inhibiting DHODH or GPX4 promotes ferroptosis in cancer cells. DHODH inhibitors may be administered to treat cancers with low expression of GPX4 as a supplement of inhibitors of system Xcor as a synergist of this inhibitor in high GPX4-expression cancers. DHODH links the mitochondrial respiratory chain with pyrimidine synthesis (Mao et al., 2021;Figure 2
).Regulators of ferroptosis
Intercellular E-cadherin-mediated contacts can regulate some ferroptosisrelated molecules, including ACSL4 and TFR, through the merlin (NF2)‒Hippo‒yes associated protein (YAP) signaling pathway axis. By antagonizing this signaling, epithelial or metastatic cancer cells resisting various treatments show high sensitivity to ferroptosis (Wu et al., 2019). In hepatocellular carcinoma (HCC), both total and nuclear YAP expression is higher than those in nontumorous regions. The density of HCC cells is related to the decreased nuclear expression of YAP and increased resistance to ferroptosis. Furthermore, epidermis-type lipoxygenase-3 is a YAP-transcriptional enhancer factor target gene promoting ferroptosis (Qin et al., 2021). S-phase kinaseassociated protein 2, an E3 ubiquitin ligase, is regulated by YAP, essentially promoting ferroptosis. Its deletion could inhibit erastin-induced ferroptosis (Yang et al., 2021). In meningioma cells, the myocyte enhancer factor 2C promotes the expression of NF2 and E-cadherin, and silencing myocyte enhancer factor 2C can enhance ferroptosis which can be limited by forcing expression of myocyte enhancer factor 2C, NF2 and E-cadherin (Bao et al., 2021b). O-linked beta-N-acetylglucosaminylation enhances and stabilize the expression of YAP, via which the susceptibility of HCC cells to ferroptosis is increased (Zhu et al., 2021a). This modification is dependent on the hexosamine biosynthesis pathway, for which glutamine-fructose-6-phosphate transaminase is the rate-limiting enzyme. Thus, in lung adenocarcinoma cells, YAP expression and O-linked beta-N-acetylglucosaminylation need the glutamine-fructose-6-phosphate transaminase. Adenylyl cyclase-mediated activation of protein kinase A can phosphorylate and inhibit glutaminefructose-6-phosphate transaminase, which is promoted by accumulated endogenous glutamate (Zhang et al., 2021d;Figure 1
).The nuclear NRF2 is an essential modulator of cellular antioxidants, and many of its targeted proteins and enzymes are responsible for inhibiting lipid peroxidation and subsequent ferroptosis via synthesis/metabolism of iron/metal and GSH (Dodson et al., 2019). Quiescin sulfhydryl oxidase 1—a cellular pro-oxidant—promotes ubiquitination-mediated degradation of epidermal growth factor receptor, thus inhibiting activation of NRF2, disrupting redox homeostasis, and sensitizing HCC cells to ferroptosis (Sun et al., 2021). Glutathione S-transferase zeta 1, an enzyme in the catabolism of phenylalanine, can suppress the level of NRF2, whose depletion increases the GPX4 level and inhibits ferroptosis, which is closely related to sorafenibresistance of hepatoma cells (Wang et al., 2021). Ubiquitin-specific-processing protease 11 deubiquitinates and stabilizes NRF2, which contributes to the suppression of ferroptosis (Meng et al., 2021). Calcium/calmodulindependent protein kinase Quiescing 2 negatively regulates ferroptosis through the AMP-activated protein kinases-NRF2 pathway in melanoma (Wang et al., 2022). Microsomal glutathione-S-transferase 1 prevents ferroptosis in pancreatic cancer cells by binding ALOX5 suppressing lipid peroxide, which is regulated by NRF2 pathway (Kuang et al., 2021). Conversely, the knockout of NRF2 significantly promotes the motor coordination of aged mice and prevents the deposition of brain iron in Parkinson’s disease and high-iron diet mouse models. Decreasing the FPN1 level of microvascular endothelial cells may hinder the process of iron entry into the brain, contributing to this phenomenon (Han et al., 2021;Figure 2
).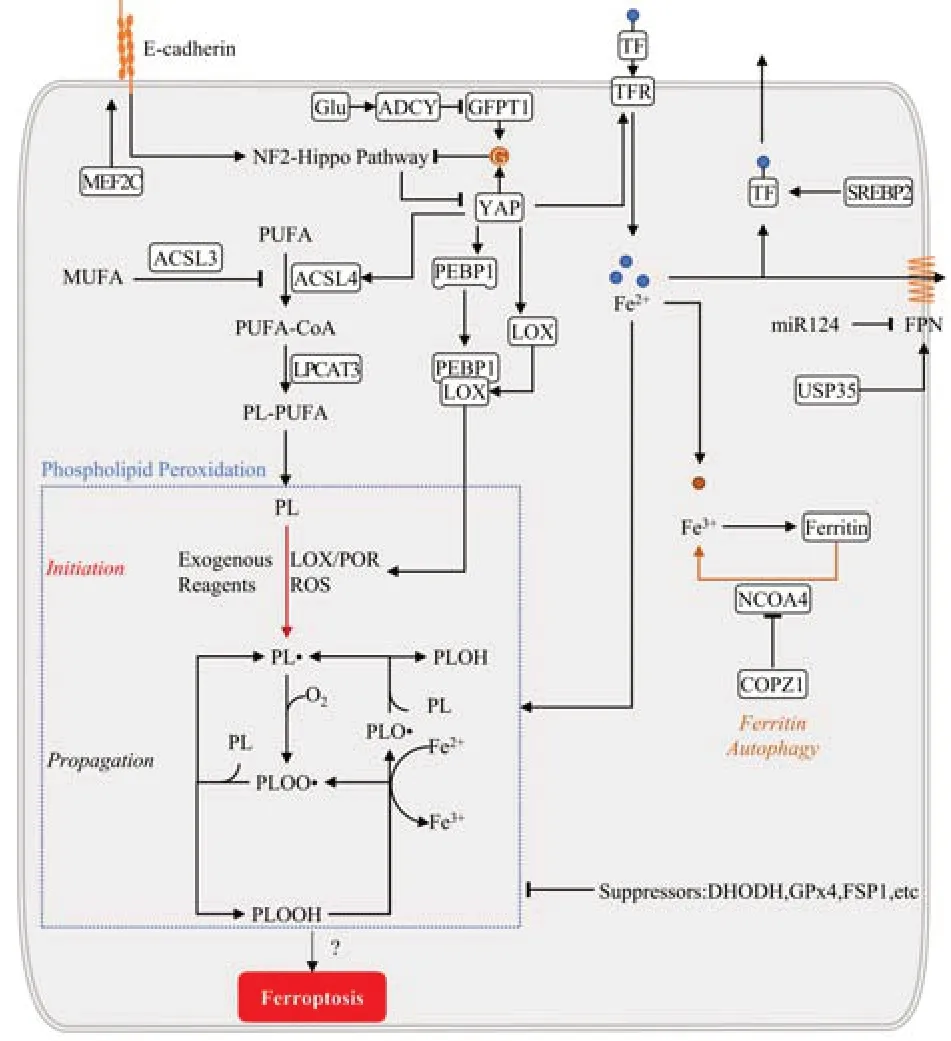
Figure 1 |Mechanism of ferroptosis. Ferroptosis is executed by iron-dependent phospholipid peroxidation. It needs processes including iron metabolism, lipogenesis, and finally lipid peroxidation. Iron metabolism relies on the transport, transition, and storage of metal iron. Iron transport into cells is through transferrin receptor (TFR), which can promote ferroptosis, and transport out of cells is via ferroprotein (FPN) and transferrin (TF), inhibiting ferroptosis. Ferritin stores Fe3+, and its autophagy can release Fe3+ and aggravate ferroptosis. Lipogenesis mainly involves producing phospholipids containing polyunsaturated fatty acid chains (PUFAPL) mediated by acyl-coenzyme A (CoA) synthetase long-chain family member 4 (ACSL4). Multiple other enzymes are required for phospholipid (PL) peroxidation and ferroptosis. Phospholipid peroxidation can be initiated by physical and chemical reagents, enzymes including lipoxygenases (LOXs), cytochrome P450 oxidoreductase (POR), and reactive oxygen species (ROS). Once the initial phospholipid hyperoxides (PLOOHs) are produced and not cleared by suppressors, activating propagation reacts with cellular labile iron to accumulate PLOOH. YAP is the crucial modulator in catalyzing ferroptosis. It is regulated by E-cadherin‒merlin (NF2)‒Hippo‒yes associated proteins (YAP) pathway and promotes iron accumulation, PUFA-PL synthesis, and phospholipid peroxidation for ferroptosis via increasing the expression of TFR, LOX, ACSL4, and others. ACSL3: Acyl-CoA Synthetase Long-Chain Family Member 3; ADCY: adenylyl cyclase; COPZ1: coatomer protein complex subunit zeta 1; G: O-glcNAcylation; GFPT1: glutamine-fructose-6-phosphate transaminase; Glu: glutamate; LPCAT3: lysophosphatidylcholine acyltransferase 3; MEF2C: myocyte enhancer factor 2C; MUFA: monounsaturated fatty acid; NCOA4: nuclear receptor coactivator 4; PEBP1: phosphatidylethanolamine binding protein 1; PL•: phospholipid radical; PLO•: phospholipid alkoxyl; PLOO•: phospholipid peroxyl radical; SREBP2: sterol regulatory element-binding transcription factor 2; USP35: ubiquitinspecific protease 35.

Figure 2 |Suppressors of ferroptosis. Glutathione peroxidase type 4 (GPX4), the canonical and most studied ferroptosiscontrolling suppressor, entails reducing with glutathione (GSH) generating oxidized glutathione (GSSG), which can be recycled via glutathione-disulfide reductase (GSR) electrons from nicotinamide adenine dinucleotide phosphate (NADPH). Ferroptosis suppressor protein 1 (FSP1) and dihydroorotate dehydrogenase (DHODH) can prevent lipid peroxidation by reducing ubiquinone (CoQ) to ubiquinol (CoQH2). Reaction for FSP1 needs electrons from NADPH and is found in the membranes of cells, Golgi complex, the nucleus. DHODH is found in the inner membrane of the mitochondria, which entails reduced flavin mononucleotide (FMNH2) to contribute electrons. Nuclear factor erythroid 2-related factor 2 (NRF2) is the core regulator of antioxidation and restrains ferroptosis. It can inhibit ferroptosis by increasing iron storage and transfer via ferritin, ferroprotein (FPN), and others; GSH synthesis by increasing the expression of glutamatecysteine ligase catalytic subunit (GCLC), GPX4, and others; and inhibiting lipid oxidation. 4EBP: Eukaryotic translation initiation factor 4E binding protein 1; A: ARE, antioxidant response element; AMPK: AMP-activated protein kinases; CAMKK2: calcium/calmodulindependent protein kinase kinase 2; EGFR: epidermal growth factor receptor; FMN: flavin mononucleotide; Glu: glutamate; GSS: glutathione synthase; GSTZ1: glutathione S-transferase zeta 1; LOX: lipoxygenase; MGST1: microsomal glutathione-S-transferase 1; mTOR: mechanistic target of rapamycin complex 1; QSOX1: quiescin sulfhydryl oxidase 1; SLC7A11: solute carrier family 7 member 11; USP11: ubiquitin-specific-processing protease 11; γGC: γ-glutamyl-cysteine; γGP: γ-glutamyl-peptides.
Ferroptosis Plays a Crucial Role following Central Nervous System Trauma
Ferroptotic changes after CNS traumatic injury
Shrunken mitochondria, the morphological feature of ferroptosis, can be observed 15 min following SCI by transmission electron microscopy and becomes more evident after 24 hours. An increase in total iron level and lipid peroxidation can be detected for 2 weeks with consumption of GPX4, SLC7A11, and GSH (Yao et al., 2019; Zhang et al., 2019; Ge et al., 2022). Ubiquitin-specific-processing protease 11 is highly expressed in neurons following hypoxia-reoxygenation, promoting autophagy by stabilizing Beclin 1, resulting in ferroptosis. Knockout ubiquitin-specific-processing protease 11 protects neurons from ferroptosis in mice with spinal cord ischemia/reperfusion injury, thus promoting functional recovery; overexpression has converse results (Rong et al., 2021).
Similar to SCI, after TBI, shrunken mitochondria can be found on the margin of the injured cortex on day 3 (Xie et al., 2019). TFR1, FPN, Fth, and ferritin L all increase post-TBI. The iron transport protein TF, TFR1, and FPN generally decrease to or fall below the average level. However, iron storage proteins Fth and ferritin L are maintained beyond the expected level for 2 weeks (Rui et al., 2021). Significant increases in Feand total iron levels can be detected in serum on day 1 post-TBI. In the ipsilateral cortex, iron, Fe, Fe, and total iron are not deposited immediately but increase within 3 days and continue to increase over time after TBI (Xie et al., 2019; Rui et al., 2021). Additionally, iron overload is detrimental to the blood-brain barrier, aggravating blood’s exudation and forming a vicious circle. Endothelial iron homeostasis contributes to blood-brain barrier integrity and stimulates hypoxia-inducible factor 2α subunit expression, preserving cadherin on the endothelial cell surface (Rand et al., 2021).
The level of lipid ROS and biomarkers of lipid peroxidation, 4-hydroxy-2-nonenal, and malondialdehyde are remarkably upregulated 3 days following TBI (Rui et al., 2021). The accumulation of ALOX15-mediated lipid oxidation products is one of the characteristics of ferroptosis and significantly oxidized phosphatidylethanolamine, which is more abundant than other PLs in the brain (Nessel and Michael-Titus, 2021). Thus, TBI quickly results in ferroptosis with an increased expression of ALOX15 and ACSL4, overload of oxidized phosphatidylethanolamine, and decrease of GSH in the ipsilateral cortex. The inhibitor of ALOX15 protects the brain against ferroptosis (Kenny et al., 2019). ACSL4 promotes neuronal ferroptosis via mediating lipid peroxidation. In acute cerebral infarction, the expression of ACSL4 is suppressed via hypoxia-inducible factor 1α subunit signaling. Knockdown of ACSL4 can inhibit microglial activation and, afterward proinflammatory cytokine production (Cui et al., 2021). The p53 protein is a critical regulator of ferroptosis. Sirtuin 2 is an NAD-dependent deacetylase that could mitigate TBI by regulating ferroptosis via p53. Inhibition of sirtuin 2 increases the expression and acetylation of p53, aggravating ferroptosis following TBI, which was significantly blocked by the knockout of p53 (Gao et al., 2021;Figure 3
).
Figure 3 |Ferroptosis-related changes and symptoms after SCI and TBI.After SCI and TBI, multiple changes associated with ferroptosis appear, including iron homeostasis dysfunction, GSH synthesis disruption, and increased phospholipid oxidation. Inflammation plays a critical role in this phase. Microglia and macrophages are transformed into the M1 state under the condition of lipid peroxidation and iron accumulation. Inflammation results in ferroptosis in local lesions and damages distant brain tissue after SCI. Similar changes occur following brain injuries. These include acute and chronic iron homeostasis dysfunction, lipid oxidation, inflammation, and tau and amyloid β (Aβ) proteins deposition. These changes are associated with acute and chronic symptoms, such as central pain (CP) and chronic traumatic encephalopathy (CTE). ACSL4: Acyl-coenzyme A synthetase long-chain family member 4; BBB: blood-brain barrier; Fbxo10: F-box protein 10; FPN: ferroprotein; GPX4: glutathione peroxidase type 4; GSH: glutathione; HIF-1A: hypoxia-inducible factor 1, α subunit; HIF2A: hypoxia-inducible factor 2, α subunit; IRE: iron-responsive-element binding protein; LOX: lipoxygenases; NF-κB: nuclear factor-κB; NOX2: nicotinamide adenine dinucleotide phosphate oxidase 2; PL: phospholipids; Prok2: prokineticin-2; PTGS: prostaglandin-endoperoxide synthase; SCI: spinal cord injury; SLC7A11: solute carrier family 7 member 11; sirt2: sirtuin 2; TBI: traumatic brain injury; TF: transferrin; TFR: transferrin receptor; TFR1: transferrin receptor 1; TNF: tumor necrosis factor.
Immunization related to ferroptosis following CNS trauma
Ferroptosis is not identified as associated with inflammation and immunization (Jiang et al., 2021). However, lipid and iron overload are commonly proinflammatory, especially in the CNS. Microglia and macrophages may polarize to a detrimental (M1) or beneficial (M2) state, mainly toward an M1 phenotype after CNS trauma. After SCI, excessive intracellular lipids and dysregulated lipid metabolism lead to the generation of foamy macrophages, which shift to the M1 phenotype. Further, myelin-activated macrophages play a crucial role in axonal retraction and dieback (Van Broeckhoven et al., 2021). Iron accumulation following SCI in macrophages can upregulate tumor necrosis factor expression, preventing M1 state skewing toward M2 polarization, appearing as a proinflammatory mixed M1/M2 phenotype. Iron overload in M2 macrophages induces a quick transformation from M2 to M1. These effects favor significant and chronic macrophage M1 polarization (Kroner et al., 2014). Furthermore, SCI-activated microglia can secrete abundant nitric oxide, which decreases the expression of ferritin and increases the expression of TFR1, divalent metal transporter 1 (DMT1), and the iron regulatory protein 1 in motor neurons. These alterations induce iron overload and increase lipid ROS production, which begets ferroptosis and primary atrophy of the motor cortex (Feng et al., 2021). Three months after TBI, macrophages containing iron and myelin degradation in the lesion area in the adjacent brain regions can be observed in mice model; however, ipsilateral and contralateral microglia and astroglia activated in the brain have a negative correlation with posttraumatic memory deficits (Lopez-Caperuchipi et al., 2021). Iron and lipid ROS accumulation and related microglia/macrophages transformation become a connection between ferroptosis and CNS inflammation.
System Xcmarkedly increased as an anti-ferroptosis factor following TBI and gradually decreased. TBI leads to decreased cortical GSH levels (Rui et al., 2021) and GPX4 activity (Xie et al., 2019). The GSH/GPX4 axis regulates LOX and prostaglandin-endoperoxide synthase (PTGS) activities. LOX and PTGS products are pro- and anti-inflammatory, modulating ferroptosis. Broken cells following ferroptosis release proinflammatory damage-associated molecular patterns which can activate the innate immune system breaking the balance between proinflammatory and anti-inflammatory action, especially in diseased kidney and brain tissues (Proneth and Conrad, 2019).
As a biomarker of ferroptosis, NADPH oxidase 2 (NOX2) expression markedly increased post-TBI in the ipsilateral cortex (Rui et al., 2021). Moreover, Nox2 and nuclear factor-κB expression maintained high levels predominantly in the microglia and macrophages at 4 and 7 days post-TBI, respectively. Inhibition of NOX2 post-TBI reduced ROS in myeloid cells. Genetic knockout or inhibition of NOX2 suppressed the proinflammatory M1 microglia/macrophages while increasing the anti-inflammatory M2 subtype in the lesion. These alterations are linked to down-regulation of the nuclear factor-κB pathway in microglia/macrophages, which can decrease proinflammatory cytokines after TBI (Wang et al., 2017). Following blast-induced TBI, expression of NOX subtypes are differently increased in various regions: It can increase NOX1 in the hippocampus and thalamus, and NOX2 in the frontal cortex. In neurons, the increase of NOX1 and NOX2 levels is higher than those in astrocytes and microglia, which indicates that neurons are more sensitive to oxidative damage than glial cells (Rama Rao et al., 2018;Figure 3
).The symptoms associated with ferroptosis
Increased iron levels occur in some brain areas after SCI through the nitric oxide synthase-iron regulatory protein 1 pathway. This intracranial iron change can activate central pain (Meng et al., 2017). Peripheral injury can generate iron-related metabolic disorders of the spinal cord and allodynia. Spinal glutamate receptor, N-methyl-D-aspartate 2B (NR2B)-containing N-methyl-D-aspartate, can elicit iron overload and mechanical allodynia. NR2B phosphorylation at Tyr1472 increases kalirin-7 expression to intensify iron accumulation and spine morphogenesis via iron-responsive-element binding protein (IRE)-DMT1 signal. Tibial fracture upregulates IRE-DMT1-mediated iron overload expression via this signal pathway and initiates lasting allodynia, which can be inhibited by deferoxamine, knockdown of kalirin-7, and antagonist of NR2B (Zhang et al., 2021a). In remifentanil-induced hyperalgesia, the level of spinal IRE-DMT1 and iron significantly increase, which N-methyl-D-aspartate receptor antagonists can also inhibit significantly (Shu et al., 2021).
Iron accumulation modulated by spinal cord N-methyl-D-aspartate modulating IRE-DMT1 plays an essential role in central pain. Acute post-TBI headache is associated with iron accumulation in multiple brain regions, proven by magnetic resonance transverse relaxation rates (T2) (Nikolova et al., 2022;Figure 3
).Tau and amyloid precursor protein are related to cellular iron retention in the brain associated with ferroptosis. Tau-mediated elimination of iron overload following ischemic stroke may prevent ferroptosis in adult mice (Tuo et al., 2017). Amyloid precursor protein can lower neuronal iron and confer neuroprotection in TBI (Ayton et al., 2014). Functional failures of these two proteins contribute to age-related, iron-modulated neurotoxicity and iron accumulation in TBI tissue. This functional deficiency may exaggerate secondary injuries after TBI. Iron accumulation and homeostasis dysfunction supposedly play a central role in mild TBI pathophysiology and its related longterm cognitive dysfunction. The prolonged cognitive impairment seems to be mediated via three pathways: iron accumulation mediates tau phosphorylation, forming neurofibrillary tangles; neuronal death; and iron accumulation results in damage of neural networks via axonal damage caused by the iron sensibility of oligodendrocytes (Huang et al., 2021b). In a study on the relationship between necroptosis and CTE, chronic brain damage could be detected almost exclusively in iron-accumulated areas and was significantly reduced in mice deleted receptor-interacting protein kinase 1 or 3 (Wehn et al., 2021).
Additionally, recent research on a large postmortem cohort proved that iron, via activating ferroptosis, may play an additional downstream role in neurodegeneration, acting independently of tau or amyloid (Ayton et al., 2021). Magnetic resonance imaging has demonstrated that regional brain iron quantification is associated with neuropsychological test scores in mild TBI; iron overload in deep gray matter—like the globus pallidus, thalamus, and hippocampus—may contribute to the pathology of TBI, eventually leading to neurological decline and cognitive impairments (Raz et al., 2011; Lu et al., 2015). Consistent with the iron deposition following TBI, iron-related proteins may contribute to long-term cellar iron pool overload, leading to ferroptosis. Glasgow coma scale scores at clinical admission and fatal outcomes after severe TBI correlate with increased serum ferritin levels (Simon et al., 2015). Thus, ferritin may be an exciting postmortem biomarker to evidence the injury pattern and survival time of fatal TBI (Ondruschka et al., 2018). By quantifying iron deposition, demyelination, and atrophy over 2 years after SCI, researchers found that cord atrophy and cerebellar loss decreased, and while brain white and gray matter atrophy was sustained, the myelin content in the spinal cord and cortex reduced progressively. As the sustained atrophy in the thalamus progressed, iron deposition was significant. Thus, myelin losses and iron deposition relate to long-term neuropathic pain intensity, motor damage, and sensory impairment and highlight the intimate relationships between degeneration of the spinal cord and brain (Ziegler et al., 2018;Figure 3
).Treatments for Ferroptosis following Central Nervous System Trauma
Recent reviews have listed some medicines for inhibiting ferroptosis after CNS traumatic injuries (Shen et al., 2020; Hu et al., 2021). In this section, we update some new studies on ferroptosis prevention and the supplements used as medicines (Additional Table 1
).Spinal cord injuries
Deferoxamine, an iron chelator, can increase the expression of GPX4, system Xc-, and GSH, inhibit gliosis, and eventually recover the long-term motor function (Yao et al., 2019). Furthermore, it can reduce motor cortex iron overload and neuronal ferroptosis following SCI and contribute to motor function recovery (Feng et al., 2021). SRS 16‒86, a novel ferroptosis inhibitor with high stability in the plasma and liver, was shown to elevate GPX4, GSH, and system Xc, reduce lipid oxidation, gliosis, and neuron injury in an SCI model (Zhang et al., 2019). Proanthocyanidin, a potent free radical scavenger commonly found in grape seeds, can decrease the levels of iron and thiobarbituric acid-reactive substances and diminish the expression of ALOX15 and ACSL4, while elevating the concentration of heme oxygenase-1 (HO-1), NRF2, GSH, and GPX4; consequently, it can rescue functionality after SCI (Zhou et al., 2020).
In one study, Fer-1, an inhibitor for ferroptosis, decreased iron and ROS deposition and downregulated the ferroptotic genes, IREB2 and PTGS2, to inhibit ferroptosis in oligodendrocytes, finally alleviating white matter injury and improving functional recovery in an SCI rat model. In addition, Fer-1 could inhibit the activation of reactive astrocytes and microglia (Ge et al., 2021). It was found that Zinc gluconate can increase GSH, superoxide dismutase, and GPX4 and decrease ROS, malondialdehyde, and lipid peroxides via increasing NRF2/HO-1 signaling pathway expression and attenuating ferroptosis in spinal cord contusion. Zinc gluconate also cured injured mitochondria and inflammation, although the NRF2 inhibitor Brusatol could reverse the effects of zinc (Ge et al., 2021). Eventually, zinc gluconate may promote behavioral and structural recovery after SCI. Lipoxin A4, an anti-inflammatory mediator, can induce neuroprotective and function improvements after SCI by regulating Akt/NRF2/HO-1 signaling (Lu et al., 2018). Subsequently, in primary spinal cord neurons via the same signaling pathway, lipoxin A4 can also inhibit erastin-induced ferroptosis (Wei et al., 2021).
TBI
In one study, Fer-1-decreased intracellular iron accumulation and the number of degenerating neurons diminished the volume of damaged lesions and ameliorated prolonged motor and cognitive prognosis after TBI in mice (Xie et al., 2019). Liproxstatin-1 (Lip-1), as a positive control of melatonin in a study by Rui et al. (2021), prevented the expression and transcription of ferroptosisrelated proteins, including system Xc, Fth, TFR1, FPN, ferritin L, Fth, and 4-hydroxy-2-nonenal following TBI. Both Lip-1 and melatonin prevented TBIinduced downregulation of cortical GSH levels and increased lipid oxidative product and iron deposition. Lip-1 and melatonin treatment attenuated neuronal death and ameliorated the motor performance (Rui et al., 2021). In oligodendrocytes, Lip-1 was shown to inhibit lipid peroxidation and restore the levels of FSP1, GPX4, and GSH, thereby ameliorating ferroptosis induced by inhibition of GPX4 which could alleviate spinal cord and brain demyelination (Fan et al., 2021).
Melatonin (N-acetyl-5-methoxytryptamine), a hormone secreted by the pineal gland and other organs, can improve TBI-induced damages and alleviate functional deficits, as mentioned above, obtaining similar effects with Lip-1. Fth-knockout mice were found to be more sensitive to ferroptosis following trauma, and the neuroprotection by melatonin was essentially canceled in Fth-knockout mice, which demonstrated neuroprotection of melatonin partly via preventing Fth-mediated neurons’ ferroptosis following TBI (Rui et al., 2021). Based on RNA sequencing, Wu et al. (2022) identified the expression of different circular RNAs after melatonin treatment for TBI and found that melatonin could exert anti-endoplasmic reticulum stress and anti-ferroptosis effects in brain injury by decreasing lipid peroxidation via the circPtpn14/miR-351-5p/5-LOX signaling. miR-212, one of the known microRNAs, is essential for synaptic plasticity, memory formation, and maintaining the blood‒brain barrier integrity in the brain. It was constantly and marked downregulated with the expression of ferroptosis-related molecules until 3 days after TBI. miR-212 can improve spatial learning and memory ability in TBI mice. Bioinformatics prediction and reverse transcriptase polymerase chain reaction verification proved that miR-212-5p attenuates ferroptotic neuronal death after TBI partly by targeting PTGS2 (Xiao et al., 2019).Baicalein, the inhibitor of 15-lipoxygenase, is known to attenuate oxidation of phosphatidylethanolamine, reduce ferroptotic protein-expression and cell death, and improve functional neuronal outcome after TBI, suggesting that it can inhibit ferroptosis and treat TBI (Kenny et al., 2019). By suppressing ferroptosis, baicalein can significantly reduce the seizure score, number of seizures, and seizure duration in FeCl3-induced post-traumatic epilepsy (Li et al., 2019). Prokineticin-2, a component of black mamba venom and frog skin, and a member of the prokineticin family, has been shown to upregulate F-box protein 10 expression and promote ACSL4 ubiquitination and degradation. Intracerebroventricular injection of Adeno-associated virus-prokineticin-2 increased Gpx4 expression, decreased the level of Acsl4 and the lesion volume, and then improved motor ability-learning performance following TBI. This prokineticin-2-driven cascade alleviates ferroptosis and protects mitochondria and neurons against injuries (Bao et al., 2021c).Polydatin, a compound extracted from Polygonum, exhibited potent neuroprotective effects in TBI via protecting against ferroptosis. It can reverse TBI-induced iron deposition, lipid peroxidation, and expression of genes correlating with ferroptotic cell death. Of note, this medicine has higher competence to improve GPx4 activity than Fer-1 (Huang et al., 2021a). Ruxolitinib, a potent and selective oral inhibitor of Janus kinases 1 and 2, was demonstrated to exert a neuroprotection effect on TBI-induced ferroptosis as efficiently as Fer-1. Ruxolitinib treatment can significantly reverse the TBI-induced lower expression of GPX4 in neurons more capably than Fer-1. Moreover, it can reduce neurodegeneration, brain edema, motor dysfunction, memory deficits, and anxiety behavior post-TBI (Chen et al., 2021).
Certain other drugs may also protect against neuronal death via inhibiting ferroptosis following TBI, but the evidence is insufficient. Tannic acid can reverse lipid peroxidation, decrease GSH level, activities of antioxidant enzymes, and the expression level of 4-hydroxy-2-nonenal. Moreover, it can effectively ameliorate the behavioral alterations, oxidative damage, mitochondrial impairment, and inflammation, which may be attributed to activation of the peroxisome proliferator-activated receptor gamma coactivator 1-α/NRF2/HO-1 signaling pathway (Salman et al., 2020). There is no evidence that it inhibits iron accumulation, but the above text has mentioned that NRF2 pathway could impact iron homeostasis and ferroptosis.Almost all selenium compounds have shown prevention of neuronal damage. They are effective against erastin- and Ras-selective lethal small molecule 3-induced ferroptosisin vitro
. In the cerebral ischemia-reperfusion injury mouse model, pretreatment with methyl selenocysteine or selenocysteine protects against neuronal damage, which supports the use of these selenium compounds for TBI treatment (Tuo et al., 2021).N,N′-di(2-hydroxy benzyl) ethylenediamine-N,N’-diacetic acid monohydrochloride, a unique iron chelator with the ability to penetrate the intact blood-brain barrier, has a higher affinity to iron and a longer half-life than most common chelators. Administration of this compound significantly reduces lipid peroxidation and motor deficits after TBI. It can decrease TBI-induced cortical injury volume, hippocampal swelling, and total hemispheric volume (Khalaf et al., 2018).Conclusion
Ferroptosis plays a crucial role in secondary injuries after CNS traumatic injuries, including free iron overload, lipid peroxidation, and ferroptosisrelated protein overexpression. These injuries can induce subsequent activation of neuroglia, immunoreaction, and inflammation, which need inhibition by treatment of ferroptosis. Researchers and clinicians have observed iron deposition, lipid accumulation, oxidation, and their related systematic changes for a period. Ferroptosis is a crucial point that connects all such secondary injuries. The new strategies for CNS treatment can reduce ferroptosis and induce cascade reactions, thus finally reaching neuron reconnection and functional reestablishment. Furthermore, many other related drugs may be used for inhibited ferroptosis, influencing iron metabolism, lipid oxidation, and redox reaction. Ferroptosis is universal in the nervous system, from neurogenesis to degeneration and from acute injury to chronic prognosis. A limitation of this review is that it did not include clinical studies. Currently, there are few clinical studies that have investigated ferroptosis. To our knowledge, the only clinical studies in this field primarily focused on biomarker identification. However, inhibition of ferroptosis has shown significant potency in protecting neurotraumatic injury in basic experiments. Further study of ferroptosis in nerve trauma will provide new avenues to clinical treatment in the future.
Author contributions:
Review conception: YJJ; literature collection and manuscript draft: QLS. Both authors contributed to manuscript revision, read, and approved the final version of the manuscript for publication.
Conflicts of interest:
The authors declare that there is no potential conflict of interest.
Availability of data and materials:
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:
Sagar Gaikwad, The University of Texas, Medical Branch at Galveston, USA; Yuri Matteo Falzone, San Raffaele Scientific Institute, Italy.
Additional files:
Effects of ferroptosis inhibitors on central nervous system injuries.
Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- Notice of Retraction
- Neuroprotective role of Noggin in spinal cord injury
- Combined cell-based therapy strategies for the treatment of Parkinson’s disease: focus on mesenchymal stromal cells
- Lights at night: does photobiomodulation improve sleep?
- β2-Microglobulin exacerbates neuroinflammation, brain damage, and cognitive impairment after stroke in rats
- The lymphatic drainage systems in the brain: a novel target for ischemic stroke?