
Ataxia-telangiectasia mutated plays an important role in cerebellar integrity and functionality
2022-11-11 05:27YuliaMitiaginAriBarzilai
中国神经再生研究(英文版) 2023年3期
Yulia Mitiagin, Ari Barzilai,
Abstract Accumulating evidence indicates that ataxia-telangiectasia mutated kinase is critical for maintaining cellular homeostasis and that it has both nuclear and cytoplasmic functions. However, the functions of ataxia-telangiectasia mutated that when lost lead to cerebellar degeneration are still unknown. In this review, we first describe the role of ataxia-telangiectasia mutated in cerebellar pathology. In addition to its canonical nuclear functions in DNA damage response circuits, ataxia-telangiectasia mutated functions in various cytoplasmic and mitochondrial processes that are critically important for cellular homeostasis. We discuss these functions with a focus on the role of ataxia-telangiectasia mutated in maintaining the homeostatic redox state. Finally, we describe the unique functions of ataxia-telangiectasia mutated in various types of neuronal and glial cells including cerebellar granule neurons, astrocytes, and microglial cells.
Key Words: ataxia telangiectasia; ATM; cerebellum; DNA damage response; double-strand breaks; mitochondrial dysfunction; oxidative stress; single-strand breaks

Introduction 497 Search Strategy and Selection Criteria 497 Ataxia-Telangiectasia 497 Cerebellar Vulnerability in Ataxia-Telangiectasia 497 ATM Is a Master Regulator of Key Nuclear and Cytoplasmic Processes 498 ATM Deletion Impairs the Redox State of the Central Nervous System 498 The Role of ATM in Tissue Regeneration 499 The Non-Nuclear Function of ATM 499 Atm Deficiency Results in Impaired Functionality of Neurons and Glial Cells 500 Future Directions 501
Introduction
DNA damage is caused in living organisms primarily by endogenous oxygen radicals produced during metabolism, which induce tens of thousands of DNA lesions per cell per day (Shiloh, 2020). Ataxia-telangiectasia mutated (ATM) is critical responses to genotoxic stresses, maintenance of the cellular redox balance, and regulation of mitochondrial metabolism (Aguado et al., 2022). When ATM is missing, as it is in ataxia-telangiectasia (A-T) patients, modulation of these pathways is defective, which results in a complex, progressive, disease. The aim of this review is to outline the current state of knowledge of the roles of ATM in tissue regeneration and in regulation of the cellular redox state and on the functions of ATM in nuclear and the nonnuclear compartments.
Search Strategy and Selection Criteria
Studies cited in this review were published from 1926 to 2022, with a predominant emphasis from 2017 to 2022 (more than 50%). All studies cited were searched on the PubMed database using the following keywords: ataxia telangiectasia, ATM, cerebellum, glia, microglia, astrocytes, granule neurons, Purkinje neurons, DNA damage response, oxidative stress, mitochondria, neural networks, and metabolism. All studies were cited due to their relevance to the review.in older patients [reviewed in (Rothblum-Oviatt et al., 2016; van Os et al., 2017; Levy and Lang, 2018; Amirifar et al., 2019)]. Growth retardation and occasional endocrine abnormalities are also seen, particularly insulin-resistant diabetes. Premature aging has recently been recognized in A-T patients (Tal et al., 2018; Aguado et al., 2022). Major laboratory findings include elevated serum levels of a-fetoprotein and carcinoembryonic antigen.
All A-T patients have mutations in theATM
gene (Savitsky et al., 1995). VariousATM
mutations that cause classical and milder forms of A-T have been identified. A-T patients show a striking sensitivity to the cytotoxic effect of ionizing radiation (IR), and ATM-deficient cells exhibit marked chromosomal instability, sensitivity to IR and radiomimetic chemicals, and reduced telomere length (Rothblum-Oviatt et al., 2016). IR sensitivity results from a profound defect in the cellular response to DNA double-strand breaks (DSBs) due to the lack of ATM protein kinase (Rothblum-Oviatt et al., 2016). Chromosomal instability observed in patients is also suggestive of defects in V(D)J recombination (Menolfi and Zha, 2020). Mouse cerebellar cells lacking Atm have also been shown to have impaired mitochondrial functionality (Stern et al., 2002). Furthermore, mammalian cells lacking Atm have increased levels of reactive oxygen species (ROS) and deletions in mitochondrial DNA in the region encoding cytochrome c oxidase (Lee and Paull, 2020). Post-mortem studies revealed a significant loss of Purkinje and granule neurons in the cerebella of children with A-T (Tal et al., 2018).Ataxia-Telangiectasia
Cerebellar Vulnerability in Ataxia-Telangiectasia
A-T (OMIN #208900) is a highly pleiotropic, autosomal recessive disorder (Rothblum-Oviatt et al., 2016) caused by mutations in the gene that encodes the ATM protein kinase (Shiloh, 2020). The first cases of A-T were documented in 1926 (Syllaba, 1926). More than four decades later, in 1958, A-T was described as a familial syndrome with progressive ataxia, oculocutaneous telangiectasia, and frequent pulmonary infection (Boder and Sedgwick, 1958). Many A-T patients develop peripheral neuropathy during the second decade of life. Oculocutaneous telangiectasia, marked immunodeficiency, neuromotor dysfunction, and gonadal and thymic dysgenesis are also typical symptoms. Cancer predisposition is manifested as an increased tendency to develop lymphoreticular malignancies, and various carcinomas appear
Accumulating evidence indicates that ATM has numerous functions in the nucleus and in the cytoplasm that help maintain cellular homeostasis. The reason for cerebellar vulnerability in A-T is still unknown (Rothblum-Oviatt et al., 2016; Shiloh, 2020; Kwak et al., 2021; Lee et al., 2021). Possibilities include its regulatory roles in the dynamics of cerebellar networks, transcription, vasculature integrity, or neural-glial-vascular-immune interactions. Despite extensive research, it is still unclear why ATM deficiency affects the cerebellum and the dopaminergic system more severely than the rest of the brain.
Ataxia is defined as a loss of coordination that in most cases affects gait and limb functions. There are at least two types of ataxias: dominant, which occurs mainly in adults and is characterized by polyglutamine accumulation (McIntosh et al., 2021), and recessive, which occurs early in life and is typified by malfunctioning DNA repair and oxidative stress (Tu et al., 2020). ATM is a major player in DSB repair; however, malfunctioning DSB repair does not specifically affect the central nervous system but rather leads to genomic instability, which can cause malignancy and immunodeficiency. Various types of cerebellar ataxias such as oculomotor apraxias AOA1 and AOA2 and spinocerebellar ataxia with axonal neuropathy (SCAN1) occur due to malfunctioning of the single-strand break (SSB) repair system (Jiang et al., 2017). ATM is activated by SSBs, coordinating their repair with DNA replication (Lee and Paull, 2021). It has been found that post-mitotic cells from patients with SCAN1 fail to repair SSBs, whereas proliferating cells were repaircompetent and appeared normal (Lee and Paull, 2021). The accumulation of SSBs in non-dividing cells such as neurons can lead to impaired transcription that can culminate in cell death. In proliferating cells, SSBs are converted to DSBs, which are then accurately repaired by homologous recombination (Wilson and Mattson, 2007). These observations led us to speculate that ATM deficiency results in the accumulation of SSBs that affect cerebellar homeostasis.
A-T is characterized by the attrition of Purkinje neurons, which are specifically cerebellar cells. The firing rate of Purkinje neurons is among the highest in the neuronal cells in the brain, and these neurons operate as a fast pacemaker with a mean half width of 0.23 ms (Masoli et al., 2015). The vulnerability of Purkinje neurons to ATM deficiency may lead to cerebellar attrition. Alternatively, ATM loss may impact other cell types that in turn cause loss of Purkinje neurons. Mutant mice have shed light on the roles of Purkinje neurons in animal behavior. Mutation in an ATP/GTP binding protein, Agtpb1, led to the loss of all Purkinje neurons within 3 weeks while sparing the granule neurons. Surprisingly, these mutant mice had only mild behavioral abnormalities (Le Marec and Lalonde, 1997). A more pronounced behavioral effect, the lurcher phenotype, was detected in theGrid2
mutant mice in which the gene encoding Grid2, a glutamate receptor, has been deleted. This mutation leads to the loss of 100% of Purkinje neurons, at least 90% of granule neurons, 30% of deep nuclei, and 60‒75% of inferior olivary neurons (60‒75%) within 2 weeks of birth (Hilber and Caston, 2001; Lalonde and Strazielle, 2007). Mice lacking Girk2, an inward rectifying K-channel, lose 40% of the Purkinje neurons and 100% of granule and dopaminergic neurons of the midbrain and have severe behavioral deficits (Lalonde and Strazielle, 2007). Kim et al. (2020) showed that loss of both Atm and Polymerase b results in severe ataxia but not Purkinje cell loss. Collectively, these results suggest that there is not a simple relationship between the degree of behavioral impairment and the severity of cell loss. For example,Grid2
- andAgtpb1
-null mice have better motor capabilities thanGirk2
mutants, despite the total absence of Purkinje neurons in theGrid2
- andAgtpb1
-null strains, suggesting that partial loss of Purkinje neurons promotes worse dysfunction than a complete absence.The average glucose consumption per neuron is nearly 20 times higher in the cerebral cortex than in the cerebellum (Herculano-Houzel, 2011). This stems from the fact that about 80% of all brain neurons are in the cerebellum, and the average glucose use per cerebellar neuron is markedly lower than that of a cortical neuron (Herculano-Houzel, 2011). The neuron to glia ratio depends on the cerebellar region [reviewed in (von Bartheld et al., 2016)]. Overall, it is 4.35:1 in the cerebellum (Herculano-Houzel, 2014). In contrast, in the cerebrum, which is mostly composed of non-neuronal cells, the neuron to glia ratio is 1:1.48 (von Bartheld et al., 2016). Astrocytes account for around 20% of all glial cells (Herculano-Houzel, 2014), and our work has shown that cerebellar network dynamics of granule neuronal cultures are heavily dependent on astrocytes (Kanner et al., 2018). Furthermore, in the mouse brain, Atm deficiency leads to age-dependent attrition of the cerebellum and dopaminergic system but not the cerebral cortex (Kirshner et al., 2012), similar to the relentless process in A-T patients.
Microglia are the resident immune cells in the brain and much mystery surrounds their function. Long viewed as the brain’s defenders against biological threats and injury, these chameleon-like cells transform from a resting to an activated, macrophage-like state when challenged by injury or disease. Microglia are rapidly recruited to sites of damage where they phagocytose debris and unwanted and dying cells. Although critical for the immune response to infection and trauma, microglia also contribute to pathological neuroinflammation by releasing cytokines and neurotoxic proteins (Levi et al., 2022). Recent data implicate microglia in many functions required to build and wire the developing central nervous system, ranging from neurogenesis to synaptic pruning (Cornell et al., 2022). Our group found that Atm deficiency results in significant impairment in cerebellar microglial function with little effect on cerebral microglia (Schlam-Babayov et al., 2021; Levi et al., 2022). TheAtm
cerebellar microglia did not survive as long and were also impaired in phagocytosis, cell migration, expression of specific proteins such as BDNF and Akt, and mitochondrial functionality compared to wild-type cells (Levi et al., 2022). The importance of fully functional microglia on neuronal survival was further demonstrated by Song et al. (2019)who showed that a conditioned medium of microglia treated with the Atm inhibitor Ku60019 caused severe structural damage to cultured neurons, most likely due to neurotoxic substances secreted by the microglia that lack Atm activity. Together, these data demonstrate the importance of microglia, and of Atm, in normal cerebellar functionality.ATM Is a Master Regulator of Key Nuclear and Cytoplasmic Processes
ATM is a serine/threonine kinase that belongs to a family of PI-3 kinaselike protein kinases (Shiloh and Ziv, 2013; Shiloh, 2020) that includes the catalytic subunit of the DNA-dependent protein kinase and ATR (Blackford and Jackson, 2017), both of which are involved in the response to genotoxic and other stresses. ATM phosphorylates more than 1000 proteins, which are involved in cell-cycle control, DNA repair, apoptosis, modulation of chromatin structure, and other cellular processes (Shiloh and Ziv, 2013; Stracker et al., 2013; Schlam-Babayov et al., 2021). A summary of ATM cellular functions is presented inFigure 1
.
Figure 1 |Cellular roles of ATM. In the central nervous system, ATM is both a nuclear as well as a cytoplasmic protein. In the nucleus, ATM plays a vital role in the DNA damage response pathway activated in response to DSBs or SSBs. ATM has an important role in maintaining a proper cellular redox state and is activated by oxidative stress. In the peroxisome, ATM regulates catalase activity and lipid peroxidation. ATM can also regulate the cellular redox state by controlling the production of the vital antioxidant NADPH via the activation of the pentose phosphate pathway. ATM also regulates the number of lysosomes, which accumulate in ATM-deficient cells. ATM regulates autophagy by the activation of the LKB1/AMPK pathway resulting in TSC2 activation, which culminates in the repression of mTORC1 activity. Aggrephagy, which prevents the accumulation of misfolded proteins, is also regulated by ATM. ATM is associated with endoplasmic reticulum (ER) stress. ATM is also implicated in the metabolism of synaptic vesicles through association with VAMP2 and SYNAPSIN-1 and with endo- as well as exocytosis via the AP-2 system. One of the most important cytoplasmic roles of ATM is the regulation of mitochondrial functionality and mitophagy. Blue arrows indicate upregulation and red arrows indicate downregulation of various molecules or processes due to ATM activity. AMPK: AMPactivated protein kinase; ATM: ataxia-telangiectasia mutated; DBS: double-strand breaks; LKB1: serine-threonine liver kinase B1; mTOR: murine target of rapamycin; NADPH: nicotinamide adenine dinucleotide phosphate; SSB: single-strand breaks; TSC2: tuberous sclerosis complex 2; VAMP2: vesicle-associated membrane protein 2.
Important downstream targets of ATM are the genome guardian p53 and the histone H2AX. p53 responds to diverse cellular stresses to regulate cellcycle arrest, senescence, DNA repair, changes in metabolism, apoptosis, and necrosis (Blackford and Jackson, 2017; Aki and Uemura, 2021). ATMdependent H2AX phosphorylation is one of the earliest signs of DNA damage. H2AX phosphorylation recruits other proteins that initiate the chromatinremodeling process essential for efficient DSB repair (Oberdoerffer and Miller, 2022).
DNA damage is caused in living organisms primarily by endogenous ROS produced during metabolism, which induces tens of thousands of DNA lesions per cell per day (Shiloh, 2020). Defense against threats to genome integrity is critical for cellular homeostasis and prevention of undue cell death, cancer, and premature aging (McKinnon, 2013; Hou et al., 2019). ATM might have an overarching role in the maintenance of genome stability as it supports numerous DNA repair pathways that respond to a variety of genotoxic stresses (Tal et al., 2018). ATM is the primary transducer of the DSB response network (Shiloh and Ziv, 2013). In response to DSBs, in a process dependent on the MRN complex, which includes Mre11, Rad50, and Nbs1, the ATM dimer is dissociated by autophosphorylation on specific sites (S367, S1893, S1981, and S1996), resulting in an activated monomeric ATM (Menolfi and Zha, 2020).
ATM Deletion Impairs the Redox State of the Central Nervous System
In 1997, Rotman and Shiloh (1997) proposed that most of the pleiotropic nature of A-T can be explained by elevated oxidative stress. In support of this hypothesis, ATM has been implicated as a key player in the maintenance of the cellular redox balance (Lee and Paull, 2021) and mitochondrial metabolism (Stern et al., 2002; Valentin-Vega et al., 2012). These functions of ATM involve different modes of ATM activation (Lee and Paull, 2021), and evidence suggests that ATM’s capacity as a protein kinase is exploited in signaling pathways in addition to those associated with DNA damage, some of which are cytoplasmic (Tal et al., 2018). Thus, when ATM is missing, as it is in A-T patients, the modulation of numerous pathways becomes suboptimal.
Oxidative stress occurs due to an imbalance between the generation of deleterious ROS or reactive nitrogen species and their neutralization by either antioxidant enzymes or low-molecular-weight antioxidants. The main antioxidant enzymes are superoxide dismutases, catalases, glutathione peroxidase, and glutathione reductase (Veal et al., 2018). Low-molecularweight antioxidants include reduced glutathione, uric acid, melatonin, and vitamin D (George and Abrahamse, 2020). ROS can be generated by the reduction of molecular oxygen to superoxide anion, hydroxyl radical, alkoxyl radicals, and peroxyl radicals. Non-radical oxidants such as hydrogen peroxide are also ROS. Reactive nitrogen species can be generated from nitric oxide metabolism. A reaction between nitric oxide and superoxide anion can generate the highly toxic peroxynitrite.
Barlow et al. (1999) have shown thatAtm
mice have abnormally high levels of superoxide anion. The mutant mice have oxidative damage to lipids and proteins in various tissues such as the brain, testes, and thymus. Levels of the inducible form of the heme oxygenase HO-1 are also detected inAtm
Purkinje neurons (Barlow et al., 1999). Kamsler et al. (2001) provided further evidence that Atm deficiency leads to oxidative stress: Levels of the sulfonated amino acid cysteine are abnormally low inAtm
brains, and activity of thioredoxin, which is a general reductase, is high in the cerebellum but not in the cerebrum of these mice (Kamsler et al., 2001). Similarly, elevated superoxide dismutase activity was detected inAtm
cerebella but not in other regions of the brain (Kamsler et al., 2001). In contrast, there was a significant decrease in catalase activity inAtm
cerebella but not the cerebra. Since superoxide dismutase converts superoxide anions to hydrogen peroxide and catalase converts it to water, this suggests that Atm deficiency specifically increases the levels of cerebellar hydrogen peroxide. High levels of hydrogen peroxide cause oxidative damage to macromolecules and also disrupt signaling pathways. This could be one cause of severe cellular dysfunction and cell death observed specifically in the cerebellum ofAtm
mice.ATM is also found in peroxisomes, which are thought to be the major sites of oxidative metabolism. Watters et al. demonstrated similarities in C-terminal sequences between ATM and catalase, which is a peroxisome-residing enzyme (Watters et al., 1999). Catalase activity is reduced in ATM-deficient fibroblasts, but the reduction in catalase activity does not stem from reduced expression of catalase, suggesting that ATM controls catalase activity thereby regulating lipid peroxidation (Watters et al., 1999).
A direct connection between the accumulation of DNA breaks and oxidative stress was demonstrated by Stern et al. (2002) who showed that Atm deficiency resulted in reductions in the reduced and the oxidized forms of NADand NADP. Pyridine nucleotides are involved in numerous essential catabolic and biosynthetic reactions in living organisms, and NADH and NADPH play critical roles in the antioxidant machinery. NADH and NADregulate the activity of PARP and SIRT2 (Covarrubias et al., 2021), factors critical in DNA repair and transcription silencing. Finally, ATM deficiency elevates the mitochondrial respiration rate specifically in the cerebellum but not in the cerebrum (Stern et al., 2002). Elevated mitochondrial respiration can lead to the generation of superoxide anions and oxidative stress. Taken together, these findings indicate that Atm inactivation can lead to unrepaired DNA breaks, which in turn impose stress on DNA damage response mechanisms. Overactivation of these systems depletes the cells of pyridine nucleotides, disrupting critical signaling pathways and cellular energetic homeostasis.
Ziv et al. (2005) generated Atm-deficient mice in the background of either Sod1 or Mlh1 deficiency. The combined deletion ofAtm
andSod1
led to increased oxidative stress, whereas combined inactivation ofAtm
andMlh1
, which encodes the mismatch repair enzyme, leading to increased genomic instability. Increasing oxidative stress exacerbated growth retardation and markedly reduced the mean survival time of mice following IR. In contrast, increasing genomic instability caused a moderate increase in radiation sensitivity but a dramatic increase in aggressive lymphomas compared toAtm
mice. These results demonstrate that Atm deficiency-induced oxidative stress and genomic instability are responsible for different phenotypes of A-T.In addition to its canonical activation, which is dependent on the MRN complex and DNA, ATM can be also activated independently of DNA damage. Using purified recombinant human ATM, Paull and her colleagues showed that ATM can be directly activated by the addition of hydrogen peroxide in the absence of the MRN complex or DNA (Lee and Paull, 2020). Under these conditions, the activated ATM is not a monomer but is a disulfide-linked covalent dimer [reviewed in (Lee and Paull, 2021)]. C2991 is necessary and sufficient to mediate ATM hydrogen peroxide-induced activation [reviewed in (Lee and Paull, 2020)]. In contrast, the positively charged amino acids R2579 and R2580 are vital for ATM activation mediated by MRN and DNA damage (Lee et al., 2018). Whereas ATM activation due to DNA damage leads to the stimulation of specific pathways responsible for survival and DNA end resection, activation of ATM by ROS regulates protein homeostasis, mitochondrial function, and ROS levels and activity. Cell-cycle checkpoint activation of various types of autophagy can be regulated either by an activated monomeric ATM or the covalent dimer (Lee et al., 2018).
The Role of ATM in Tissue Regeneration
Is there a connection between genome instability, DNA damage, and tissue regeneration? The answer is yes. Wound healing requires the migration of stem cells to the injury site where they participate in the repair process. A unique model system to study tissue regeneration is the flatworm planarian, which is capable of regenerating new tissue at the sites of injury. Moreover, planarian can also remodel existing tissue to restore a fully functional tissue. The regeneration process involves the migration of specific stem cells called neoblasts. Neoblast migration involves nuclear changes that are associated with elevated DNA damage. Upon their arrival at the injury site, the level of the DNA damage is reduced (Reddien, 2018). When DNA damage is induced prior to the injury, migration is delayed, and the sensitivity of the stem cells to further DNA damage is increased compared to stationary neoblasts. Depletion of essential components of the DNA repair system revealed that normal activation of the DNA repair machinery is vital for proper cell migration in planarian (Sahu et al., 2021). Depletion of Atm during normal regeneration in planarian did not affect the regenerative process; however, depletion of Rad51, which is a key player in the repair of DNA through homologous recombination, led to the demise of the planarians. Interestingly, depletion of Atm after sublethal IR hampered the ability of the stem cells to repopulate and to promote the regenerative process. Together, these results suggest that Atm is necessary for tissue regeneration in those cells that were exposed to DNA damage but that it does not play a role in normal migration-induced DNA damage (Sahu et al., 2021).
The Non-Nuclear Function of ATM
ATM has non-nuclear activities in different cellular domains and organelles. In neuronal cells, and especially Purkinje neurons, of mice, Atm is predominantly located in the cytoplasm (Barlow et al., 2000). Dar et al. (2006) examined Atm subcellular localization in mouse cerebellar cells using cerebellar organotypic cultures that were exposed to different doses of IR. IR results in DNA damage via its ability to cause radiolysis of the cellular water molecules thereby increasing the formation of ROS (Nuszkiewicz et al., 2020). The newly generated ROS are capable of reacting with cellular macromolecules such as lipids, proteins, and DNA resulting in damage in both the nucleus and the cytoplasm (Juan et al., 2021). Atm activation by IR is mediated by the autophosphorylation at S1987 in mice (S1981 in human) (Alexander and Walker, 2010). Phosphorylated Atm was exclusively detected in the nuclei of Purkinje neurons as well as in the nuclei of other cerebellar cells upon IR (Dar et al., 2006). If ROS-induced Atm activation is indeed cytoplasmic, phosphorylated Atm should have been detected in the cytoplasm, but it was not. It is possible that in cerebellar cells S1987 autophosphorylation occurs predominantly in the nucleus, whereas other autophosphorylation sites or covalent Atm dimerization can take place in the cytoplasm. It is also possible that only some ROS such as hydrogen peroxide results in cytoplasmic activation of Atm. This notion is supported by the fact that mitochondriaderived hydrogen peroxide promoted ATM dimerization and activation (Zhang et al., 2018).
There is also evidence that cytosolic ATM is involved in the general recycling processes known as autophagy. ATM is implicated in the clearance of damaged mitochondria and peroxisomes in response to increased oxidative stress [reviewed in (Blignaut et al., 2021)]. Under conditions of oxidative stress, ATM is activated and triggers the activity of the LKB1/AMPK pathway, which leads to activation of TSC2 in the cytoplasm. The activation of TSC2 represses mTORC1 and thereby induces autophagy. ATM can also suppress TORC1-mediated signaling pathways in response to hypoxia through the phosphorylation of HIF1a (Cam et al., 2010). ATM also mediates mitophagy, a mitochondrial degradation pathway (Liu et al., 2020). Spermidine-induced mitochondrial depolarization activates ATM leading to the recruitment of a PTEN-induced kinase PINK1 and the E3 ligase Parkin to induce mitophagy (Sarkar et al., 2021). A mutated ATM protein that is incapable of activation by oxidative stress led to widespread cellular protein aggregation (Lee et al., 2018), thereby connecting ATM and aggrephagy, which removes misfolded proteins. The accumulation of dysfunctional misfolded proteins can severely disrupt cellular homeostasis. Misfolded proteins are usually degraded by the ubiquitin proteasome system or by the lysosomal system using aggrephagyspecific chaperons (Blignaut et al., 2021). Data also implicate ATM in vesicular transport. Lim et al. (1998) demonstrated that ATM binds to β-Adaptin, which is a subunit of AP-adaptor, and β-NAP, the neuronal homolog of yeast β-Adaptin.
Atm deficiency significantly reduces hippocampal long-term potentiation. Using spontaneous synaptic dye release, Li et al. (2009) showed that Atm can form a complex with two presynaptic vesicular proteins Vamp2 (also known as synaptobrevin) and Synapsin-1. Vamp2 is associated with neurotransmitter release between docking and fusion whereas Synapsin-1 is implicated in synaptogenesis and also in synaptic transmission (Li et al., 2009). Further strengthening the notion that Atm is associated with presynaptic failure, Vail et al. reported that Atm-deficient mice failed to generate normal long-term potentiation and that Atm was associated with the presynaptic protein Piccolo but not with the postsynaptic marker Homer (Vail et al., 2016). Atm was also shown to be directly involved in the development of the GABAergic system. Reduced Atm levels or inhibition of its kinase activity by its specific inhibitor KU55933 resulted in hippocampal hyperexcitability via overexpression of Mecp2, which is involved in DNA methylation and thereby controlling gene expression (Pizzamiglio et al., 2016, 2021).
ATR, like ATM, is a central player in DNA damage response; ATR mainly functions in the repair of SSBs and resolution of stalled replication forks [reviewed in (Blackford and Jackson, 2017)]. Like ATM, ATR is also detected in the cytoplasm, most notably in neurons. ATM and ATR have both been localized to cytoplasmic vesicular structures, including synaptic vesicles where they associate with the synaptic vesicle proteins VAMP2 and SYNAPSIN-1 (Li et al., 2009). ATM associates with excitatory, glutamatergic vesicles, whereas ATR associates with inhibitory, GABAergic vesicles (Cheng et al., 2018). The complementarity also extends to their pathways of degradation. ATM and ATR are both large proteins (3056 and 2644 amino acids, respectively) with similar three-dimensional structures. Yet ATM is degraded primarily through the autophagy pathway, whereas ATR is degraded primarily by the proteosome (Cheng et al., 2020). The large sizes of ATM and ATR raise the intriguing possibility that these proteins can serve as scaffolds for complex protein assemblies. Support for this notion was recently provided by Hotokezaka et al. (2020) who showed that ATM is involved in cell death triggered by the accumulation of unfolded or misfolded proteins. The authors found that cytoplasmic ATM serves as a “platform, not a kinase” on which the phosphatase PP2A dephosphorylates AKT. ATK dephosphorylation leads to the activation of the glycogen synthase kinase GSK3b resulting in the degradation of the nascent polypeptide-associated complex of a subunit as well as g-taxilin, which accumulates due to mitochondrial-dependent cell death in response to endoplasmic reticulum stress.
Reduced mitochondrial functionality was detected inAtm
cells as well as in cells treated with small-molecule inhibitors of ATM. ATM is detected in mitochondria and is activated by mitochondrial dysfunction (Lee and Paull, 2020). Atm deficiency in thymocytes leads to increased levels of ROS, to upregulation of the respiratory capacity, and to abnormal mitophagy (Valentin-Vega et al., 2012; Tal et al., 2018). Lee et al. (2018) have shown that cells expressing an oxidation-deficient form of ATM (C2991L) have reduced mitochondrial membrane potential and mitophagy compared to wild-type cells. ATM senses redox state through the formation of ROS-dependent disulfidelinked dimers (Lee et al., 2018). ATM can also control the cellular redox state by upregulating the pentose phosphate pathway, which plays a key role in the production of the crucial antioxidant NADPH. Activated ATM phosphorylates HSP27, which binds to and activates the dehydrogenase G6PD. G6PD is rate limiting enzyme of the pentose phosphate pathway, and its activation elevates the production of NADPH. Interestingly, activation of the pentose phosphate pathway by ATM also increases nucleotide production. Like ATM-deficient cells,G6PD
cells are impaired in DSB repair (Cosentino et al., 2011).ATM deficiency leads to increased mitochondrial mass as well as a reduction in the production of ATP (Valentin-Vega et al., 2012; Levi et al., 2022), suggesting that ATM directly regulates mitochondrial functionality (Valentin-Vega et al., 2012). To regulate mitochondrial homeostasis, ATM phosphorylates the transcription factor NRF1, enhancing its ability to form dimers and its nuclear translocation. In the nucleus, NRF1 upregulates nuclear-encoded mitochondrial genes thereby increasing the activity of the electron transport chain. In mice, Atm deficiency reduces the ability of mitochondria to synthesize ATP and thus diminishes cellular homeostasis (Chow et al., 2019). These results led Chow et al. (2019) to suggest that Atm functions as a guardian of mitochondrial output. Low levels of cellular NADand mitochondrial dysfunction were detected inAtm
neurons derived from mice andC. elegans
(Stern et al., 2002; Fang et al., 2016). Replenishment of NADlevels stimulated neuronal DNA repair and improved mitochondrial functionality via mitophagy (Fang et al., 2016). This treatment also extended the lifespan and motricity in both worm and mouse models. Fang and Bohr suggested that NADserves as a signaling molecule between nuclear DNA integrity and mitochondrial functionality, two factors that are critically important for aging (Fang et al., 2016).Atm Deficiency Results in Impaired Functionality of Neurons and Glial Cells
Levine-Small et al. (2011) recorded the activity of multiple neurons and measured the firing rates of individual neurons, spike signals of individual neurons, inter-spike intervals, and the raw spike signals of individual neurons in cortical neural cultures grown from WT andAtm
mice seeded on a microelectrode array. The firing of individual neurons and the number of spikes were not affected either by Atm deficiency or by DNA damage. Moreover, no differences in cortical neural network activities were detected between wild-type andAtm
neural-glial cultures. Similar to wild-type cultures,Atm
cultures had the ability to synchronize. Careful analyses revealed that upon DNA damage theAtm
networks lost their ability to maintain synchronization persistence over time, instead of displaying sustained synchronization after DNA damage (Levine-Small et al., 2011). Kanner et al. (2018) found that Atm regulates the dynamics of cerebellar networks. The number of global synchronization events, in which most if not all the cultured cells fire together, was significantly reduced inAtm
networks, and the number of sparse synchronization events, in which a few cells fire and most cells remain silent, were significantly increased inAtm
cerebellar cultures compared to wild-type cultures. It is important to note that these differences were detected in untreated cerebellar neural-glial cultures. Remarkably, a chimeric culture of wild-type astrocytes andAtm
neurons had network dynamics and numbers of synapses at wild-type levels. In contrast, no neuronal survival or activity was detected in chimeric cultures of wildtype neurons andAtm
astrocytes (Kanner et al., 2018). Furthermore, Atm deficiency severely hampers the complexity of the Bergmann glia and velate astrocytes in the whole cerebellum. Surprisingly, Atm deficiency increased the number of excitatory synapses in the cerebellar cultures, whereas in the whole cerebellum the level of inhibitory synapses was upregulated. Astrocytes are known to affect synapse formation, maturation, and elimination (Kanner et al., 2018). Excessive numbers of synapses inAtm
cultures and cerebella may stem from increased formation or decreased elimination. We found that Atm deficiency led to increased levels of the autophagy markers p70S6K and p62 both in cultures and whole cerebella (Kanner et al., 2018). ATM negatively regulates mTOR thereby enhancing autophagy (Shiloh and Ziv, 2013), and the increased levels of both p70S6K and the p62 are correlated with inhibited autophagy. Thus, we concluded that the increased number of synapses inAtm
cultures and cerebella are most likely due to reduced autophagy. Taken together, these findings demonstrate the vital role that astrocytes play in neuronal homeostasis and in the dynamics of brain networks.Our group has shown that astrocyte function is impaired inAtm
mice, which also have severely impaired retinal vascularization and leaky blood vessels (Raz-Prag et al., 2011). These findings highlight the role of Atm in maintaining the integrity and the functionality of the neuro-glia-vascularimmune unit. Characterization of the retina in young and aged Atm-deficient mice revealed that, at 2 months of age, angiography was slightly impaired inAtm
vasculature compared to wild-type controls. Astrocytes and endothelial cell labeling demonstrated diminution of astrocyte coverage of the vessels inAtm
retinas. Atm loss also significantly reduced the complexity of the processes of astrocytes, which likely results in the observed atrophied structural organization of the astrocytic network. These results suggest that impaired vascularization in the central nervous system plays an important role in the etiology of A-T, and vascular abnormalities may underlie or aggravate brain degeneration.Human and rodent neurons are similar (Hodge et al., 2019), and the fundamental difference between the human and the rodent brain lies in the number and complexity of the glial cells (Oberheim et al., 2006). A comparison of the properties of humans (Zhang et al., 2016) and mouse astrocytes reveals that mouse astrocytes have higher tricarboxylic acid cycle activity, reduced lactate production, higher resistance to ROS, and higher expression of catalase (Ye Zhang, University of California, Los Angeles personal communication). Since astrocytes play such an important role in brain functionality as well as in brain degenerative diseases (Oberheim et al., 2006; Acioglu et al., 2021), it is our notion that highly durable and resistant mouse astrocytes ameliorate the toxic effects of Atm deficiency. This is the reason that Atm deficiency in mice does not completely mimic the effects of ATM loss in humans.
Further evidence that astrocytes play a role in the etiology of A-T was provided by Petersen et al. (2012) who showed thatAtm
knockout inDrosophila
glial cells but not in neuronal cells was sufficient to cause the A-T symptoms in flies and that ATM deficiency in astrocytes is sufficient to activate an immune response that leads to neurodegeneration. Petersen et al. (2012) suggested that non-cell-autonomous mechanisms are responsible for neurodegeneration in A-T. Conceptually similar findings have now been demonstrated by the Herrup and co-workers who discovered that ATM deficiency in microglial cells leads to enhanced DNA damage and the accumulation of genomic DNA fragments in the cytoplasm. This last alteration leads to the activation of the cGAS/STING pathway and the initiation of a robust innate immune response. The resulting cocktail of chemokines and cytokines possesses significant neurotoxic properties. Thus, as with the astrocytes, loss of ATM activity in a non-neuronal cell type is sufficient to trigger a dramatic change of state in the neurons of the brain. These findings and the work of others (Kanner et al., 2018) strengthen the notion that astrocytes are involved in almost every disease of the brain. In addition to astrocytes and microglia, ATM deficiency also affects the integrity and the functionality of oligodendrocytes leading to impaired myelination both in humans and mice (K. Herrup, personal communication). In summary, these data demonstrate that ATM plays a role in the functionality of glial cells that are deeply involved in the etiology of A-T.Figure 2
depicts the roles of ATM in various cerebellar cells and functions.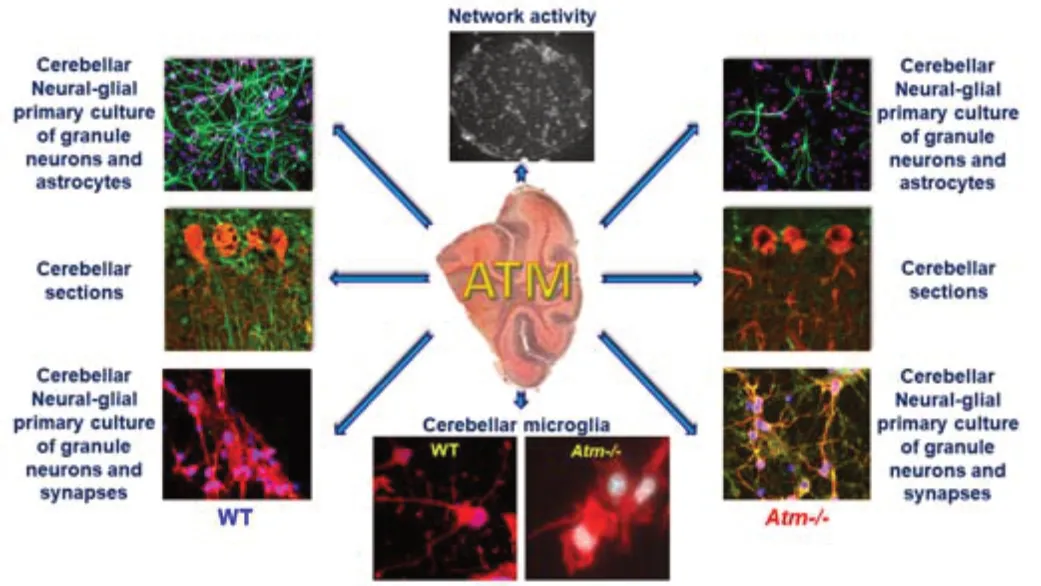
Figure 2 |Roles of Atm in whole cerebellum and cerebellar neural-glial primary cultures. In the cerebellum, Atm regulates the morphology of astrocytes. The complexity of the astrocytic processes is significantly reduced in the absence of Atm. Astrocytes play a major role in the dynamics of cerebellar network dynamics. Atm deficiency activates cerebellar microglia and alters their functionality. Atm also controls the number of synapses both in primary cultures of granule neurons and in the whole cerebellum. Further, Atm inactivation significantly hampers the dynamics of granule neuronal networks. ATM: Ataxia-telangiectasia mutated; Atm-/-: Atm-deficient mice; WT: wild type.
Future Directions
A-T is an incurable, multisystem disease that affects neural, glial, vascular, and immune system function in the cerebellum. The most devastating symptom of A-T is cerebellar ataxia, which severely disables young A-T patients, leaving them wheelchair-bound for the rest of their lives. A-T research uses many types of different cells as model systems to understand the role of ATM in cell functionality and homeostasis. These include ATM-deficient human fibroblast and lymphoblast cell lines as well as neuronal-like cell lines isolated from different animal models of A-T. In addition, some studies utilize primary cultures of cerebral cortical neurons or different cerebellar primary cultures of neurons and glial cells. The interpretations of the results obtained from thein vitro
systems must be validatedin vivo
.The heterogeneity observed in the model systems demonstrates the diverse functions of ATM. Extrapolation from one model system to another is highly problematic. Therefore, studying the appropriate model system is critically important for the generation of data relevant to the design of A-T treatments. In particular, understanding the role of each cerebellar cell in the etiology of A-T is critically important. The immediate cure for A-T would be a genetic therapy in which the mutatedATM
gene is replaced by a wild-type copy of the gene in all cells. At present, this type of therapy is not available. Thus, more pragmatic treatments should be tested.Author contributions:
Both authors equally contributed to the design and the writing of the manuscript, to data search and analysis, and to the collection of the relevant references. Both authors approved the final version of the manuscript.
Conflicts of interest:
The authors declare no conflicts of interest.
Availability of data and materials:
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:
This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:
Mark G. Clemens, University of North Carolina at Charlotte, USA.
Additional file:
Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- Notice of Retraction
- Neuroprotective role of Noggin in spinal cord injury
- Combined cell-based therapy strategies for the treatment of Parkinson’s disease: focus on mesenchymal stromal cells
- Lights at night: does photobiomodulation improve sleep?
- β2-Microglobulin exacerbates neuroinflammation, brain damage, and cognitive impairment after stroke in rats
- The lymphatic drainage systems in the brain: a novel target for ischemic stroke?