
Schimke免疫—骨发育不良1例
2022-11-07 02:45李倪
罕少疾病杂志 2022年10期
李 倪
汕头大学医学院第一附属医院全科医学分院放射科 (广东 汕头 515041)
1 一般情况
患儿10岁,因“确诊矮小症6年,发现蛋白尿2月”于2021年1月13日来我院儿童生长发育门诊就诊。患儿曾于2015年至2017年间因身材矮小在深圳儿童医院予生长激素治疗,过后无定期随诊,患儿系第一胎,其母诉怀孕时体健、孕期检查无异常,足月顺产,出生时为低体重儿(不足5斤),父、母亲平素身体健康,后育有妹妹7岁,弟弟4岁,均体健,家族无遗传病史。患儿近2年偶有双侧眼睑浮肿,数日后可自行消退,否认肢体水肿,否认有泡沫尿、血尿,否认尿频,偶有头痛,诉颞侧为主要,持续约1~2分钟可以自行缓解。患儿5天前有咳嗽,伴流黄色脓涕,无发热,无气促,无恶心、呕吐,其母诉患儿平素体质较差,多次因上呼吸道感染急诊。
2 本次就诊情况
2.1 体格检查身高130cm,体重27kg。神智清楚,反应可。发育迟缓,营养差,头颅前后径较长,下颌较短小,腰椎前凸,宽低鼻梁。胸廓正常,呼吸音粗,未闻及干湿性啰音。腹软、膨隆,腹壁无明显水肿,无压痛及反跳痛,未及包块,肝、脾肋下未触及,Murphy征阴性,移动性浊音阴性,双下肢未见明确凹陷性水肿。双上臂伸侧可见色素脱失斑。
2.2 实验室检查甲状腺功能检查示:TT3:1.63nmol/L,FT3 5.73pmol/L,TT4:143.18nmol/L,FT4 13.20pmol/L,TSH7.719mIU/L;促肾上腺皮质激素:1.05pmol/L;血脂7项:总胆固醇17.18mmol/L,甘油三酯1.69mmol/L,高密度脂蛋白胆固醇2.81mmol/L,低密度脂蛋白胆固醇11.26mmol/L;ANA+ANA谱11项:抗核抗体1:100(参考值<1:32),余阴性;抗双链DNA测定抗体2.07IU/ml(参考值0—107IU/ml);免疫球蛋白G3.67g/L(参考值7.51—15.6q/L);尿常规:蛋白质3+,潜血及白细胞阴性,RBC 50.3×106/L,尿蛋白量(24 h)5.86g。凝血功能:栓溶二聚体1620ug/LFEU(参考值<550ug/LFEU);血常规:Hb 126 g/L,Pit 175×109/L,WBC 5.68×109/L;血生化:白蛋白19.88 g/L(参考值40~55 g/L),总蛋白48.66 g/L(参考值65~85 g/L)。肾功能:Ser 42.84umol/L,BUN 2.44 mmol/L。电解质:Na 137.75mmol/L.K4.93 mmol/L,Cl106.56 mmol/L,Ca12.06 mmol/L。传染病:乙肝,丙肝,梅毒,HIV均阴性。
2.3 影像学检查(见图1~图2):骨盆X光片:双侧髂骨变短,髂骨翼变小,髋臼顶水平状,双侧股骨头细小,双侧股骨近端明显变短,双侧髋关节呈半脱位,双侧股骨头稍向外上方移位,并假关节形成。脊柱X光片:胸、腰椎椎体普遍稍变扁,侧位显示部分锥体前中部稍厚而膨隆,后部变薄且稍凹陷,状如梨形,部分锥体前缘不规则。垂体MRI:垂体MRI平扫未见明显异常;扫描野鼻咽顶后壁增厚,拟未退化腺样体;拟全组副鼻窦炎。肾脏B超:肝胆胰脾未见明显异常,右肾区未见明显肾脏回声,左肾区考虑重复肾可能,右侧隐睾,左侧睾丸未见明确异常。
2.4 2021年2月23日送检全外显子示主要及次要检测结果如下表(见表1~表2)。主要检测结果:Schimke型免疫—骨发育不良。次要检测结果:胱氨酸尿症,智力障碍合并视网膜色素变性,常染色体隐性耳聋84B型,着色性干皮病互补组D,毛发低硫营养不良1型,脑—眼—面—骨骼综合征2。

表1 全外显子示主要检测结果
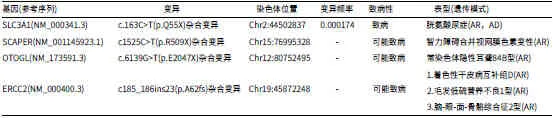
表2 全外显子示次要检测结果
2.5 染色体核型分析(见图3):核型46,X,inv(Y)(p11q11),此核型中Y染色体发生倒位。
3 讨 论
SIOD十分罕见,SIOD的主要特征是脊椎骨骺发育不良、细胞免疫缺陷、淋巴细胞减少和进行性肾功能不全[1]。除上述特征性表现外,本病还可有多种伴随症状,常见的有面部畸形、皮肤色素斑、甲状腺功能低下、发作性脑缺血等。M. Morimoto[2]等人还发现在已发现的双等位基因SMARCAL1突变的SIOD患者中有牙齿异常。几乎所有的SIOD患者均有肾脏损害,表现为不同程度的蛋白尿,其中大部分表现为肾病综合征,患者均进行性进展至肾衰竭。分析此例患儿特点如下:(1)该患儿具备Schimke免疫—骨发育不良典型临床表现,患儿出现特殊面容:身材矮小,营养不良,头颅前后径较长,下颌较短小,腰椎前凸,宽低鼻梁。双上臂伸侧可见色素脱失斑。(2)患儿影像学表现符合脊柱骨骺发育不良(SED),SED系一组选择性累及脊柱和长管状骨骨骺的染色体遗传性发育障碍疾病,临床上有躯干与肢体不成比例的矮小身材的特征,主要表现为:短躯干型侏儒症,普遍性脊柱变形,长骨骨骺骨化延迟,伴不同程度髋内翻。Schimke免疫—骨发育不良的影像学表现常与年龄有关,主要为脊柱骨骺发育不良,脊柱骨骺发育不良根据发病时间分为先天型和晚发型[3],先天型较晚发型严重,各个年龄段的椎体呈卵圆形,梨形椎体,随年龄增长,椎体普遍变平,上下板略不规则,随年龄增加,可有明显的进行性骨量减少,椎体受压,椎体骨密度与软组织相似。骨盆正位片:股骨骨骺、髂骨发育不全伴髋臼向上倾斜和成形不良。颅骨:可显示明显增宽的蝶鞍或蝶鞍前部凹陷,亦可显示正常的蝶鞍[4]。影像学上需重点与以下疾病鉴别:黏多糖贮积症IV型,常累及全身所有骨关节,亦有广泛的扁平椎,椎体前中部呈鸟嘴状,小或无枢椎齿突,尿中含有硫酸角质可明确诊断。多发骨骺发育不良(MED):常为常染色体显性遗传,特点为骨骺延迟骨化或不规则骨化从而导致髋内翻及膝内外翻,椎体累及多而轻。假性软骨发育不良:是异染色体遗传的骨骼发育不良,导致侏儒症,畸形直到幼儿期才显现,干骺端模糊,骨骺形态多样,比较罕见。(3)临床上SIOD可分为婴儿期和幼年期,婴儿型首先出现在子宫内,其特征是生长迟缓、甲状腺功能减退、骨髓衰竭、短暂性脑缺血发作、中风和肾功能衰竭,导致出生后5年内死亡。幼年期发病源于至少一个由于缺失、无意义或移码突变引起的SMARCAL1空等位基因,SMARCAL1基因的无义、帧移位和剪接突变破坏了SNF2蛋白的正常结构,从而产生截断的蛋白产物[5],幼年发病形式源于SMARCAL1等位基因的错义突变,导致SMARCAL1蛋白的功能故障,大量的双等位基因错义突变与轻微的临床症状有关,如果对终末期肾病(ESRD)进行治疗,可以活到生命的第二和第三个十年。SIOD表型由轻到重不等[6-7],轻度SIOD患者经合理治疗[8]可存活至成年,严重表型导致青少年患者死亡,最终发展为终末期肾病。
本例患者一开始因生长发育矮小而就诊,因父母家族无遗传病史,故诊疗过程未引起足够重视,此后患儿逐渐出现蛋白尿、水肿而就诊,经基因检测而确诊为SIOD,而SMARCAL1基因型[9-10]不能预测疾病的严重程度或结局,该患儿伴发低蛋白血症、大量蛋白尿及高脂血症,符合肾病综合征诊断标准,SIOD的肾脏疾病是SIOD的另一个临床显著特征,可能是通过基因表达的改变引起的[11]。从目前文献来看,SIOD预后不佳,临床对其认识不足,容易发生漏诊,多数患者晚期死于肾功能衰竭或者其他并发症,如反复感染、中风、脑梗死等。
猜你喜欢
医学美学美容(2021年21期)2021-11-20
中华骨与关节外科杂志(2021年12期)2021-08-31
中华骨与关节外科杂志(2021年12期)2021-08-31
中老年保健(2021年2期)2021-08-22
昆明医科大学学报(2021年2期)2021-03-29
新疆大学学报(自然科学版)(中英文)(2020年2期)2020-07-25
医学新知(2019年4期)2020-01-02
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
中学生理科应试(2016年4期)2016-11-19
