Notch1在新生期小鼠肺炎链球菌感染诱导Th1/Th2失衡
2022-11-07 10:32:24王艺颖张瀚之李媛媛陈诗懿张光莉罗征秀
第三军医大学学报 2022年21期
王艺颖,简 鼎,朱 琳,张瀚之,李媛媛,陈诗懿,张光莉,罗征秀
400014 重庆,重庆医科大学附属儿童医院呼吸科,国家儿童健康与疾病临床医学研究中心,儿童发育与疾病教育部重点实验室,儿科学重庆市重点实验室, 儿童感染免疫重庆市重点实验室
肺炎链球菌是儿童社区获得性呼吸道感染的第一位细菌病原体。流行病学资料表明,出生后早期呼吸道肺炎链球菌感染增加后续喘息性疾病发病风险[1-2]。我们前期研究发现,新生期感染肺炎链球菌可致Balb/c小鼠Th1/Th2 失衡,诱导气道高反应性(airway hyperresponsiveness,AHR)形成和哮喘发生[3-4]。Th1/Th2失衡通过其相关细胞因子水平变化对外周免疫反应进行调节,在气道炎症、气道重塑形成中起着重要作用[5-6],但新生期肺炎链球菌感染诱导Th1/Th2失衡的机制并未完全清楚。
Notch信号是一种进化高度保守的细胞间信号级联,由4种不同的Notch跨膜受体(Notch1、Notch2、Notch3、Notch4)和5种配体组成,Notch受体与配体结合后被γ分泌酶裂解,导致Notch受体胞内结构域断裂诱导下游效应物转移[7]。体内研究发现Notch1可促进GATA3基因表达,诱导Th2细胞分化;同时还可降低T-bet表达,抑制Th1细胞分化,诱导Th1/Th2失衡形成[8-9]。Th1/Th2失衡哮喘小鼠模型Notch1蛋白水平明显增加,利用γ分泌酶抑制剂{N-[N-(3,5-Difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester,DAPT}抑制Notch1信号通路可降低哮喘小鼠肺部Th2类细胞因子产生,纠正Th1/Th2失衡,减轻气道炎症和AHR[10-11]。新生期肺炎链球菌感染诱导Th1/Th2失衡及AHR形成是否与Notch1信号活性有关尚不清楚。因此,本研究在前期研究基础上进一步探讨Notch1信号通路对新生期小鼠肺炎链球菌感染Th1/Th2水平及AHR的影响,以期阐明新生期肺炎链球菌感染引起Th1/Th2水平失衡促进哮喘发生的可能机制。
1 材料与方法
1.1 实验动物
清洁级孕晚期Balb/c雌鼠3只,购自重庆医科大学实验动物中心,饲养于独立换气系统,空气过滤,饲料、饮用水及垫料均消毒后定期于超净台内进行更换,恒定室温(22~25 ℃)、湿度(50%~65%),光照10~12 h/d。饲料和饲养水均经过消毒处理,实验操作均在超净工作台内进行。
1.2 实验动物模型建立
孕晚期Balb/c雌鼠生产新生小鼠12只,分为3组:对照组、新生期肺炎链球菌肺炎(Streptococcuspneumoniaepneumonia,S.pp)组、新生期肺炎链球菌肺炎Notch1抑制(S.pp+DAPT)组。采用1周龄的Balb/c新生小鼠建立非致死性肺炎链球菌肺炎模型,参照课题组前期研究方法并进一步改进[12-14],即鼻腔缓慢滴注肺炎链球菌悬液1×107cfu/10 μL,对照组同期鼻腔滴注无菌PBS 10 μL。根据课题组前期结果:新生期Balb/c小鼠感染肺炎链球菌后7 d肺部细菌已被清除[15],在小鼠肺炎链球菌感染后第7天,S.pp+DAPT组按小鼠体质量鼻腔滴注DAPT(0.03 mg/kg,MedChem Express, USA)抑制Notch1信号[10],S.pp组和对照组同期鼻腔滴注等量无菌PBS。在小鼠6周龄(进入成年期)收集各组小鼠标本进行相关检测,肺炎链球菌标准株D39由重庆医科大学医学检验、临床检验诊断学教育部重点实验室惠赠。
1.3 小鼠气道反应性测定
各组小鼠6周龄时,利用不同浓度乙酰甲胆碱溶液(0、3.125、6.25、12.5、25、50 mg/mL)雾化3 min,以激发小鼠气道高反应,休息2 min后,采用法国EMKA体积描记法肺功能仪检测小鼠气道反应性[15]。
1.4 流式细胞术检测肺组织Th1、Th2水平
实验方法参照本课题组前期研究[3],气道反应性检测后置于笼内休息3~6 h,各组小鼠用过量水合氯醛麻醉处死,取全肺组织剪碎,制成单细胞悬液,孵箱孵育,加入荧光标记抗体Hamster anti-mouse CD3e- Pecy7(1∶100,BD Biosciences,USA)、Rat anti-mouse CD8a-FITC(1∶100,BD Biosciences,USA)避光孵育,加Rat anti-mouse IL-4-APC(1∶100,BD Biosciences,USA)、Rat anti-mouse IFN-γ-PerCPcy5.5(1∶100,BD Biosciences,USA)室温避光孵育,加入200 μL PBS 混匀,于流式细胞仪(FACSClibur, BD Biosciences)上机检测。
1.5 支气管肺泡灌洗液(bronchoalveolar lavage fluid,BALF)标本收集及检测
参照本课题组前期研究方法收集BALF[16],6周龄小鼠用过量水合氯醛处死后,立即行气管插管,夹闭左主支气管后,对右肺进行无菌PBS液支气管肺泡灌洗,0.5 mL /次,共3次(总回收率>80%),离心后下层细胞沉渣涂片行瑞姬氏染液染色,油镜下观察细胞形态,分类计数中性粒细胞、淋巴细胞、单核细胞和嗜酸性粒细胞数量。
1.6 Western blot检测小鼠肺组织中Notch1蛋白表达
取小鼠右肺冻存于-80 ℃冰箱内,取肺组织约100 mg,用含有1% PMSF的RIPA研磨提取全蛋白,按30 μg/孔上样量在8% SDS-PAGE中分离,采用半干法转染将蛋白转至PVDF膜上,快速封闭液封闭,4 ℃分别孵育一抗Notch1兔单克隆抗体(1∶1 000,zenbio,China)及β-actin鼠多克隆抗体(1∶1 000,北京中杉金桥)过夜,孵育二抗1 h,采用ECL进行显影,采用Image Lab软件统计目的条带灰度值。
1.7 肺组织炎症病理学检查
取小鼠完整左肺,先置于4%多聚甲醛内浸泡固定,后梯度酒精脱水、石蜡包埋,沿肺组织横切面石蜡切片(厚度为4 μm),烤箱烘干后二甲苯脱蜡,HE染色,再经酒精、二甲苯脱水透明,用树胶封片,光镜下采集图像,参照文献[17-18]对气管细支气管周围炎症、血管周围炎症和肺间质炎症进行组织炎症病理半定量评分。
1.8 RT-PCR检测小鼠肺组织中T-bet、GATA3 mRNA表达
取-80 ℃冰箱内冻存小鼠右肺组织,获取匀浆后采用总RNA提取试剂盒(Solarbio,China)提取总RNA,以总RNA中的mRNA作为模板,采用反转录试剂盒(TaKaRa,Japan)合成cDNA,以cDNA为模板进行目的基因和内参基因GAPDH的PCR扩增,经过变性、退火、延伸后检测每组小鼠肺组织中T-bet、GATA3的表达水平。基因引物序列为:T-bet正向引物5′-GCCTACCAGAACGCAGAGAT-3′,反向引物 5′-AGC-AGTTGACAGTTGGGTCC-3′;GATA3正向引物5′-CTC-GGCCATTCGTACATGGAA-3′,反向引物 5′-GGATAC-CTCTGCACCGTAGC-3′;GAPDH正向引物5′-CAGCGA-CACCCACTCCTCCACCTT-3′,反向引物5′-CATGAG-GTCCACCACCCTGTTGCT-3′。
1.9 统计学分析
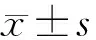
2 结果
2.1 鼻腔滴注DAPT抑制新生期肺炎链球菌肺炎后小鼠肺组织Notch1蛋白表达
Western blot结果显示,S.pp组小鼠发育至成年期,肺组织中Notch1蛋白表达水平显著高于对照组(P<0.05,图1)。S.pp+DAPT组小鼠发育至成年期时,肺组织Notch1蛋白水平较S.pp组、对照组均显著降低(P<0.01,图1)。提示新生期肺炎链球菌感染促进肺组织Notch1蛋白表达,鼻腔滴注DAPT能成功抑制肺炎链球菌感染后Notch1蛋白表达。
2.2 阻断Notch1对新生期肺炎链球菌肺炎后小鼠BALF细胞分类计数的影响
收集各组小鼠BALF行细胞分类计数(图2)。结果显示:S.pp组小鼠细胞总数、中性粒细胞、巨噬细胞、淋巴细胞计数均显著高于对照组。S.pp+DAPT组小鼠BALF细胞总数、中性粒细胞、单核细胞、淋巴细胞计数均明显低于S.pp组(P<0.01)。提示阻断Notch1能明显减少新生期肺炎链球菌肺炎后小鼠气道炎症细胞浸润。
a:P<0.01,与对照组比较;b:P<0.01,与S.pp组比较
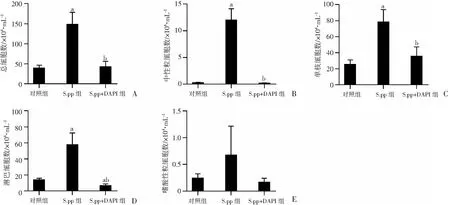
2.3 阻断Notch1对新生期肺炎链球菌肺炎后小鼠肺组织病理的影响
HE染色观察各组小鼠肺组织病理情况及组织炎症病理评分(图3)。结果显示:S.pp组气管细支气管周围炎症、血管周围炎症及肺间质炎症评分均显著高于对照组(P<0.01);S.pp+DAPT组气管细支气管周围炎症、血管周围炎症及肺间质炎症评分显著低于S.pp组(P<0.01),与对照组比较差异无统计学意义。提示本研究建立的非致死性新生期肺炎链球菌肺炎小鼠模型成功,同时显示阻断Notch1能减轻新生期肺炎链球菌肺炎后小鼠肺组织炎症。
a:P<0.01,与对照组比较;b:P<0.01,与S.pp组比较
2.4 阻断Notch1对新生期肺炎链球菌肺炎后小鼠肺组织T-bet、GATA3 mRNA表达的影响
RT-PCR检测各组小鼠肺组织T-bet、GATA3 mRNA的表达水平(图4)。结果显示:S.pp组T-betmRNA表达显著低于对照组(P<0.01),GATA3 mRNA表达明显高于对照组(P<0.01);S.pp+DAPT组T-betmRNA表达与S.pp组比较差异无统计学意义,但显著低于对照组(P<0.01),GATA3 mRNA表达在S.pp+DAPT组明显低于S.pp组(P<0.01),但明显高于对照组(P<0.05)。提示阻断Notch1能明显抑制新生期肺炎链球菌肺炎后小鼠肺组织GATA3 mRNA表达。

a:P<0.05,b:P<0.01,与对照组比较;c:P<0.01,与S.pp组比较
2.5 阻断Notch1对新生期肺炎链球菌肺炎后小鼠肺组织Th1、Th2细胞水平的影响
流式细胞术检测各组小鼠肺组织中Th1、Th2细胞水平并分析Th1/Th2比值(图5)。结果显示:S.pp组Th1细胞水平显著低于对照组,Th2细胞水平明显高于对照组,Th1/Th2比值显著低于对照组,差异均有统计学意义(P<0.01)。阻断Notch1表达后,S.pp+DAPT组小鼠肺组织Th1细胞水平与S.pp组比较差异无统计学意义,但显著低于对照组(P<0.01);S.pp+DAPT组小鼠肺组织Th2细胞水平明显低于S.pp组(P<0.01),而与对照组比较差异无统计学意义;S.pp+DAPT组小鼠肺组织Th1/Th2比值较S.pp组显著增加(P<0.01)。提示阻断Notch1能显著抑制新生期肺炎链球菌肺炎后小鼠肺组织Th2细胞表达,部分逆转Th1/Th2失衡。
2.6 阻断Notch1对新生期肺炎链球菌肺炎后小鼠气道反应性的影响
体积描记法检测各组小鼠雾化吸入不同浓度乙酰甲胆碱后的气道反应性(图6)。结果显示:吸入乙酰甲胆碱浓度为3.125、6.25、12.5、25、50 mg/mL时,S.pp组小鼠气道反应性较对照组均显著增高;阻断Notch1后S.pp+DAPT组小鼠吸入上述浓度乙酰甲胆碱的气道反应性较S.pp组明显降低,S.pp+DAPT组小鼠吸入乙酰甲胆碱浓度为12.5、25、50 mg/mL 时气道反应性明显高于对照组,在乙酰甲胆碱较低浓度3.125、6.25 mg/mL时差异无统计学意义。提示阻断Notch1能明显降低新生期肺炎链球菌肺炎后小鼠气道高反应性。
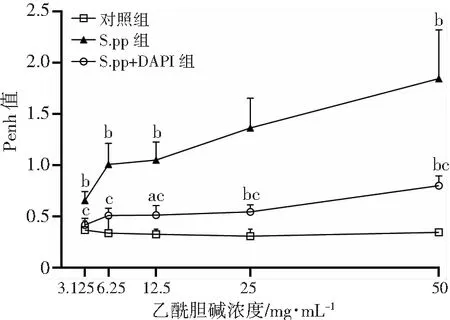
a:P<0.05,b:P<0.01,与对照组比较;c:P<0.01,与S.pp组比较
3 讨论
哮喘是一种以慢性气道炎症、气道高反应性为特征的异质性疾病[19]。尽管哮喘病因及发病机制尚未完全清楚,但Th1/Th2失衡是公认的哮喘发生机制[10]。Th1和Th2细胞是由Th0发育而来的CD4+T细胞亚群,生理状态下,Th1/Th2可维持动态平衡,异常刺激可诱导Th1/Th2失衡,进而诱导免疫异常相关疾病发生[20]。我们前期研究结果显示,新生期Balb/c小鼠感染肺炎链球菌诱导成年期肺组织Th1/Th2失衡,促进AHR及慢性气道炎症形成[3]。
Notch信号通路是一种高度进化保守的细胞信号通路,调节胚胎和成人组织细胞增殖、分化,与AHR形成和炎症细胞分化密切相关[11]。动物实验研究发现RSV感染诱导AHR与慢性气道炎症形成,伴随小鼠肺部Notch1水平显著增加[21]。Notch1通路经γ分泌酶水解后引起下游效应,本研究发现新生期感染肺炎链球菌后,Balb/c小鼠肺部Notch1水平显著增加,小鼠气道反应性明显增加、气道炎症细胞浸润显著增多;而感染后鼻腔滴注DAPT阻断Notch1表达,小鼠吸入不同浓度乙酰甲胆碱后气道反应性均较感染组明显降低,气道炎症细胞浸润显著减少,提示新生期肺炎链球菌感染诱导AHR形成及气道炎症细胞浸润与Notch1活化相关。
体内研究发现Notch1与初始CD4+T细胞、树突状细胞等细胞表面不同配体结合后,在IL-4、IFN-γ等不同细胞因子影响下,诱导T-bet、GATA3等不同转录因子表达,最终诱导不同CD4+T细胞亚型分化而发挥作用[8,22]。Th1和Th2效应CD4+T细胞分化分别由转录因子T-bet和GATA3调控。临床及动物实验显示,Th1/Th2平衡紊乱可诱导气道炎症、粘蛋白高分泌、AHR和气道重塑[21,23-24]。动物实验发现Notch1对CD4+Th1和CD4+Th2细胞分化具有重要作用[25-26]。本研究发现新生期肺炎链球菌肺炎小鼠肺部T-betmRNA和Th1细胞显著减少,GATA3 mRNA和Th2细胞显著增加,新生期肺炎链球菌感染诱导Th1/Th2失衡;DAPT阻断Notch1通路后,小鼠肺组织T-betmRNA和Th1细胞水平无显著改变,而GATA3 mRNA和Th2细胞显著减少,Th1/Th2失衡部分改善。提示新生期肺炎链球菌感染后阻断Notch1信号主要抑制小鼠肺部CD4+Th2细胞表达,部分逆转Th1/Th2失衡,改善AHR及气道炎症细胞浸润。阻断Notch1信号后Th1/Th2未完全恢复正常水平,可能与该感染模型对Th2分化影响更显著以及其他因子如Runx、IRF4、STAT等参与Th1、Th2细胞分化有关[27]。
综上所述,新生期小鼠肺炎链球菌肺炎激活Notch1信号通路诱导肺部Th1/Th2失衡,抑制Notch1表达可部分逆转Th1/Th2失衡,减轻气道高反应性及气道炎症细胞浸润。
猜你喜欢
中国慈善家(2022年1期)2022-02-22 21:39:45
皮肤病与性病(2021年3期)2021-07-30 08:07:30
国际呼吸杂志(2019年22期)2019-12-09 09:20:26
国际呼吸杂志(2019年5期)2019-03-30 01:38:20
国际呼吸杂志(2019年3期)2019-03-01 05:39:06
国际呼吸杂志(2019年2期)2019-02-14 06:11:26
海峡姐妹(2017年7期)2017-07-31 19:08:23
中国医药生物技术(2015年4期)2015-12-26 08:26:36
视野(2015年4期)2015-07-26 02:56:52
中国记者(2014年1期)2014-03-01 01:37:29