
诺氟沙星环己甲基衍生物铜配合物的合成研究★
2022-11-06 11:05:20汪铃泓邢迦迦
山西化工 2022年7期
王 曼,汪铃泓,邢迦迦
(太原工业学院化学与化工系,山西 太原 030008)
引言
喹诺酮类药物是一类新型的合成抗菌药,它具有较广的抗菌谱和较强的抗菌作用[1]。然而,随着研究的深入,科学家们也发现喹诺酮类药物有许多副作用和耐药性,氟喹诺酮类药物还存在一些其他缺点,如生物利用度低、血药浓度低,以及其他一些影响疗效的因素[2-4]。因此,人们希望合成具有稳定构型和新的医疗功能的喹诺酮类药物。
诺氟沙星是第三代喹诺酮药物,它是一种抗菌谱广、抗菌活性强的合成抗菌药物,对大多数细菌都有明显的抑制作用。在研究过程中发现,如果在哌嗪基上的N-4’位上接入烷基,不仅能够增强对革兰氏阳性菌的抑制作用,同时,引入烷基(脂溶性基团)后,还可以增加药物的脂溶性,改善药物在体内的疗效[5-6]。
目前,金属配合物的研究也正在兴起中,喹诺酮类药物在体内的活性也会受金属离子影响。若在喹诺酮类药物的3,4-位羰基上引入吸电基团,增强电负性,易与金属离子发生配合作用,形成的金属配合物以插入和表面结合沟槽的形式与细菌DNA 相互作用,阻止细菌生长,在抗菌和抗癌物质中发挥良好作用。查阅文献[7]发现,目前对诺氟沙星衍生物的合成或者其配合物的合成研究已十分广泛,而对于其衍生物金属配合物的合成研究相对空白,本文在根据现有文献研究报道的基础上,选择诺氟沙星环己甲基衍生物作为喹诺酮药物分子的基础配合物,侧重研究合成Cu2+离子的诺氟沙星环己甲基衍生物的铜配合物,探索新的抗菌活性和药代动力学性质。并且探索合成诺氟沙星环己甲基衍生物铜配合物的最佳工艺条件和工艺路线,表征其产品的结构[8-9]。
1 实验部分
1.1 实验原理
1.1.1 诺氟沙星环己甲基衍生物的合成反应原理
本实验合成诺氟沙星环己甲基衍生物的反应原理是N 烷基化反应原理,见图1。反应使用诺氟沙星、溴代甲基环己烷作为主反应原料,诺氟沙星N-离子作为亲核试剂进攻溴代甲基环己烷的碳原子,整个反应属于SN2亲核取代反应。反应过程中,在哌嗪环的N 上形成新的C-N 键,N-4’位上接入环己甲基。
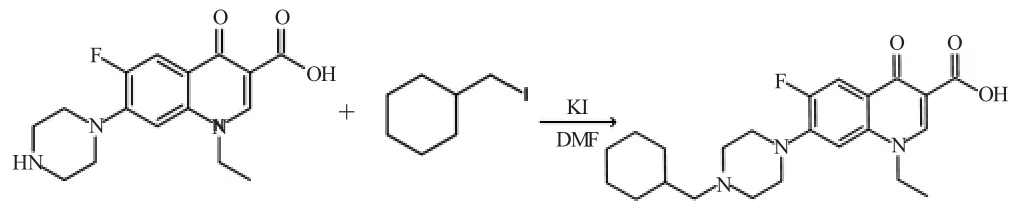
图1 诺氟沙星环己甲基化衍生物的合成反应式
1.1.2 诺氟沙星环己甲基衍生物的铜配合物的反应原理
在诺氟沙星环己甲基衍生物的3,4 位羰基上引入吸电子基团即环己甲基,导致其电负性增强,易与金属离子发生配位反应,产生金属配合物,见图2。该配合物的作用机制是细菌DNA 螺旋酶的抑制剂。
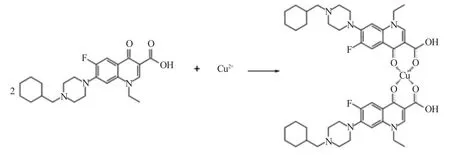
图2 诺氟沙星环己甲基衍生物铜配合物的合成反应式
1.2 实验操作步骤
1.2.1 诺氟沙星环己甲基化衍生物的合成
1)按n(诺氟沙星)∶n(溴代甲基环己烷)=1∶1.1,在分析天平上称取诺氟沙星原料药6.38 g、碘化钾3.32 g、溴代甲基环己烷3.54 g,用量筒量取DMF 30 mL加入三颈烧瓶中,加热至80 ℃,加入3.32 g 碘化钾和3.54 g 溴代甲基环己烷,反应2 h。
2)反应完成之后,把反应后的溶液转移到旋转蒸发仪中,调节温度至80 ℃,若反应到此时回收瓶内无DMF 滴入时,立即停止旋蒸。
3)将用蒸馏水洗下的固体倒入烧杯中,可以观察到溶液呈乳白色悬浊液,将装有溶液的烧杯置于磁力搅拌装置上打浆20 min,目的是充分除去KI 和DMF。
4)本实验采用二次抽滤,即抽滤-重结晶(无水甲醇)-抽滤,得衍生物产品。
5)干燥8 h 后从恒温干燥箱中取出产品,产品呈白色粉末状。用熔点法和红外光谱表征分析产品,粗略计算产率。
1.2.2 诺氟沙星环己甲基衍生物铜配合物的合成
1)称取诺氟沙星环己甲基衍生物8.32 g 和无水碳酸氢钠0.336 g,置于盛有100 mL 蒸馏水的烧杯中,加热搅拌至65 ℃,反应至溶液清亮透明。
2)用已经配好的0.1 mol/L 的NaOH 溶液调节溶液的pH 保持在7~8 之间。
3)称取4.27 g 的Cu(NO3)2·3H2O 用少量水溶解,将2)中溶液缓慢滴入铜溶液中,滴加过程中不停的搅拌,观察到会有沉淀生成。
4)室温放置到第二天进行过滤,用蒸馏水洗涤、抽滤、干燥,通过称量计算产品的产率。
5)对产品进行结构表征。
2 结果与讨论
2.1 单因素实验
2.1.1 反应温度的选择
温度影响有机合成反应程度及化学平衡。因此,为了找到最佳反应温度,需要进行单因素研究。设定反应时间为2 h,反应物料比n(诺氟沙星)∶n(溴代甲基环己烷)=1∶1.1,相转移催化剂1.0 g、碘化钾3.32 g,通过阶梯改变反应温度,得到不同温度下产品的产率,寻找产率最大时的反应温度,即最适反应温度。实验结果如下表1 所示。

表1 反应温度与产率的单因素表
通过分析图3 可知,在此折线图中,温度升高,反应收率先升后降。反应温度达80 ℃时,曲线达到最高点,即产品的产率最高;越过最高点之后,继续升高反应温度,对产率的提高也没有帮助,反而会使产率下降。因此,合成衍生物的最适反应温度为80 ℃。

图3 反应温度与产率折线图
2.1.2 反应时间的选择
反应时间也会影响反应程度。反应时间过短,反应不完全;反应时间过长,会导致反应产物的分解或副产物的生成。在合成诺氟沙星环己甲基衍生物的实验中,只改变反应时间,其他条件保持不变。实验结果如表2 所示。

表2 反应时间与产率的单因素表
在图4 中,产物收率随着反应时间的增加先上升后下降。最高点时,反应产率最高(84.3%)时的反应时间为2 h。当经过2 h 之后,增加反应时间,产率反而开始急剧下降。因此,可以得出,时间过长或者过短都不利于反应的发生,即衍生物合成的最佳反应时间为2 h。

图4 反应时间与产率折线图
2.1.3 反应物料比的选择
原料配比也是工艺研究的一个重要指标。为了找出合成反应的最佳原料配比,必须进行单因素实验。原料配比选择不当会导致反应物转化不完全,因此反应的产率不符合预期。由于反应物中的溴具有挥发性,因此反应中会发生损失。因此,有必要调整原料配比,以获得最合适的原料配比,即最佳反应物料比。通过单因素实验,固定反应的时长为2 h、反应的温度为80 ℃。每次加入相转移催化剂、活化剂KI、无水碳酸钾的质量保持不变,实验结果如表3 所示。
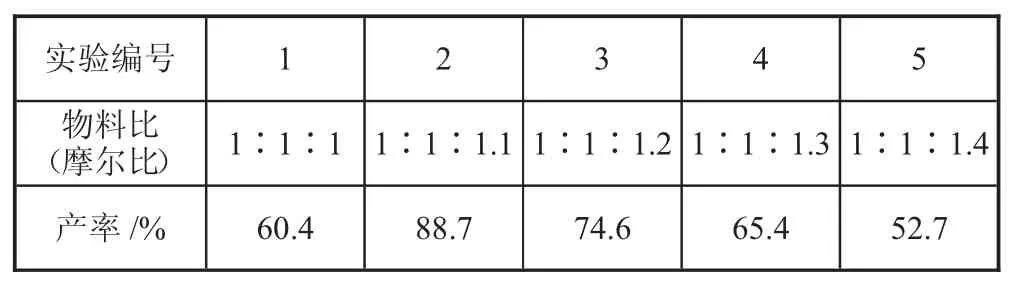
表3 反应物料比与产率的单因素实验表
由图5 可知,随着物料比中溴代甲基环己烷的量的增加,反应产率也随之增加。当反应物料摩尔比为1∶1.1 时,产率为88.7%,达到最高。继续增加物料比,反应产率又迅速下降。因此可以得出,合成诺氟沙星环己甲基衍生物的最佳反应物料比为n(诺氟沙星)∶n(溴化甲基环己烷)=1∶1.1。

图5 反应物料比与产率折线图
2.1.4 相转移催化剂对产率的影响
诺氟沙星和溴代甲基环己烷的极性大不相同。在反应过程中,两个主要原料所处的相不同。因此反应原料之间无法充分接触,使得整个反应不能完全反应或反应速度缓慢。此时,如果加入相转移催化剂,使诺氟沙星转入有机相,就可以与溴代甲基环己烷充分接触。在上述最佳条件下进行两组实验,通过比对两组实验产率,分析催化剂对反应的影响。实验结果如下表4 所示。
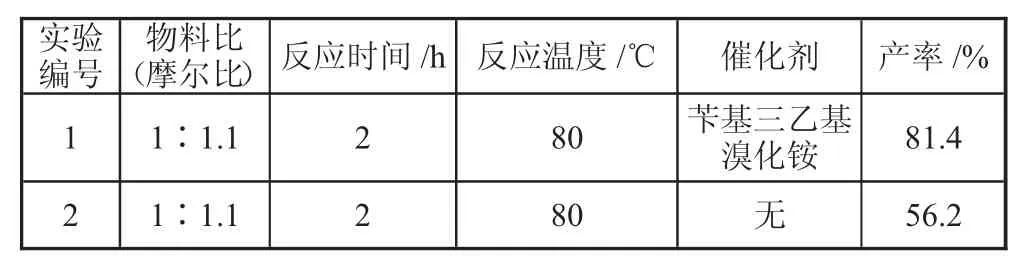
表4 相转移催化剂与产率的单因素实验表
由表4 可以看出,加入相转移催化剂的实验组产率与未加入相转移催化剂的对照组产率相差25.2%,说明加入相转移催化剂可以提高产率,并且提高的程度很大。
2.2 响应面分析
在单因素实验的基础上,选择三因素(反应时间、反应温度、反应物料比)三水平进行响应面实验。根据软件拟合给出的条件,一共设计了17 组实验,进行一系列实验操作后,实验结果如表5 所示。

表5 诺氟沙星环己甲基衍生物响应面数据
响应面软件中得到的等高线图和三维图如图6所示。

图6 等高线图和三维图
在三维图中,凸面上有一最高点,此顶点为某个因素固定时,3 个因素拟合得到的产率最高点,这个点表示合成衍生物各个因素的最佳条件,如表6 所示。

表6 环己甲基衍生物响应面最佳条件数据
2.3 重复性实验
重复性实验是指在响应面软件设计得出衍生物合成的最佳工艺条件下,再进行实验操作,已经确定的诺氟沙星环己甲基衍生物的最佳反应条件为:物料比n(诺氟沙星)∶n(溴化甲基环己烷)=1∶1.1,反应时间温度为80 ℃,反应时间为2 h。实验结果如表7所示。
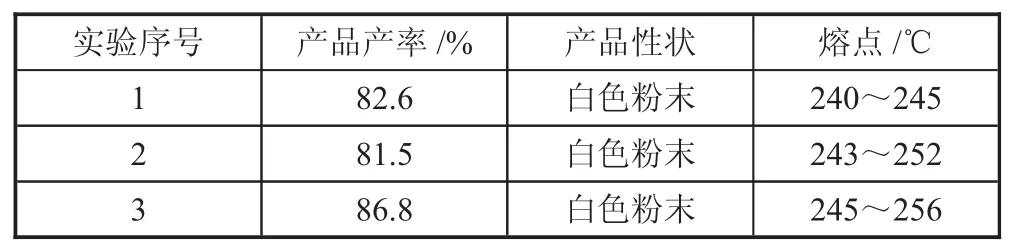
表7 重复性实验数据
由表7 中实验结果可以得出,根据最佳条件下反应的产物产率计算平均值,得出结果为83.6%。这个结果与响应面法软件拟合得出的产物产率相近,因此,可以得出,响应面软件拟合出得出的反应条件就是合成诺氟沙星环己甲基衍生物的最佳反应条件。
2.4 产品的表征
2.4.1 熔点的测定
由熔点测定仪测得诺氟沙星的熔点为218 ℃~224 ℃,诺氟沙星环己甲基衍生物的熔点为205 ℃~212 ℃,诺氟沙星环己甲基衍生物铜配合物的熔点为243 ℃~251 ℃,相比于原料药诺氟沙星,衍生物的熔点、配合物的熔点有明显改变,说明有新的物质生成。
2.4.2 红外光谱的测定
诺氟沙星的红外光谱图如图7 所示。

图7 诺氟沙星的红外谱图
诺氟沙星环己甲基衍生物的红外光谱图如图8所示。
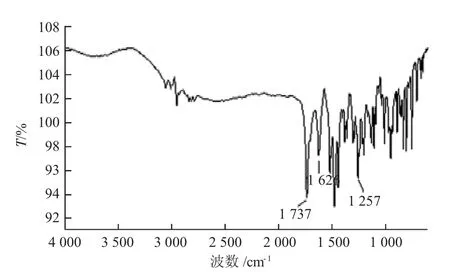
图8 诺氟沙星环己甲基衍生物的红外谱图
对诺氟沙星的红外谱图(图6)进行分析:3 320 m-1处为N-H 键吸收峰,1 726 m-1处为羧基的吸收峰,1 614 m-1处为酮羰基吸收峰。对诺氟沙星环己甲基衍生物的红外谱图(图7)进行分析:3 320 m-1处为N-H键峰消失,说明诺氟沙星的N-H 键断裂,环己甲基取代H 原子连接在N-4′位上;1 379 cm-1处为甲基振动峰,没有出现偶合情况,说明了诺氟沙星哌嗪环的N-H 键已断裂,形成了C-N 键,确定生成了诺氟沙环己甲基衍生物。
2.4.3 核磁共振氢谱的测定
图9 为诺氟沙星环己甲基衍生物的核磁共振氢谱图。从图9 中可以看出,δ 值约为3.6 ppm 处为NRC6H5基团,说明哌嗪环N-4’位上的H 已被NRC6H5取代。即确实有诺氟沙星环己甲基衍生物的生成,而且生成的产物纯度还比较高。

图9 诺氟沙星环己甲基衍生物氢谱图
2.5 电导率的测定
以二氯亚砜为溶剂,配制浓度1×10-3mol/L 的溶液。在上述溶液中测得衍生物的电导率为36.7 S·cm2·mol-1,诺氟沙星环己甲基铜配合物的电导率为44.5 S·cm2·mol-1,诺氟沙星原料药的电导率为36.2 S·cm2·mol-1,三者电导率的不同,说明有新的药物合成。
3 结论
通过合成、表征、单因素实验、响应面设计、重复性实验等一系列操作,合成诺氟沙星环己甲基衍生物的最佳工艺条件为反应物料比n(诺氟沙星)∶n(溴代甲基环己烷)=1∶1.1,反应温度为80 ℃,反应时间为2 h。由于实验时间的限制,查阅文献得合成诺氟沙星环己甲基衍生物铜配合物的最佳反应条件为:n(诺氟沙星环己甲基衍生物)∶n(三水合硝酸铜)=2∶1.1,反应溶剂为水(100 mL),反应温度是65 ℃,在此条件下产物产率最高。在表征过程中,通过熔点测定,测得诺氟沙星原料药的熔点为218 ℃~224 ℃,诺氟沙星环己甲基衍生物的熔点205 ℃~212 ℃,诺氟沙星环己甲基衍生物铜配合物的熔点为243 ℃~251 ℃,三者熔点有明显差异,说明有新物质生成。在红外光谱测定中,3 320 cm-1处为N-H 键峰消失,说明该键断裂;1 379 cm-1处为甲基振动峰,没有出现偶合情况,说明了哌嗪环的N-H 键上的H 已经被环己甲基基团取代。在核磁共振氢谱中,δ 值约为3.6 ppm 处为NRC6H5基团。以上表征手段确认了生成物的结构。
猜你喜欢
口腔护理用品工业(2021年4期)2021-11-02 08:23:00
昆明医科大学学报(2021年8期)2021-08-13 08:59:14
化工管理(2021年7期)2021-05-13 00:45:22
环境保护与循环经济(2020年4期)2020-06-08 10:43:48
中国特种设备安全(2019年1期)2019-03-13 01:06:28
保健与生活(2019年1期)2019-01-13 13:54:39
中学生数理化·高二版(2016年3期)2016-12-26 09:36:58
合成化学(2015年10期)2016-01-17 08:56:26
应用化工(2014年1期)2014-08-16 13:34:08
华东师范大学学报(自然科学版)(2014年1期)2014-04-16 02:54:58
