
基于高通量测序技术鉴定松辽黑猪与长白猪背最长肌差异基因
2022-11-03 11:54:36侍玉梅陈少康赵延辉原佳妮盛熙晖齐晓龙倪和民王楚端
华北农学报 2022年5期
侍玉梅,陈少康,邢 凯,赵延辉,原佳妮,盛熙晖,齐晓龙,倪和民,郭 勇,王楚端
(1.北京农学院 动物科学技术学院,北京 102206;2.北京市畜牧总站,北京 100101;3.中国农业大学 动物科学技术学院,北京 100193)
肌肉生长和脂肪沉积是养猪生产中非常重要的经济性状,同时也是复杂的数量性状,受到众多因素的影响[1];然而,猪肌肉生长和脂肪沉积过程中的分子调控机制仍有待进一步研究。猪的脂肪性状直接影响猪肉品质,而对不同品种猪的肉质进行比较分析是鉴定差异表达基因(DEGs)的主要方法之一。松辽黑猪作为我国培育的地方品种,具有繁殖率高、肉质优良、适应性强、耐粗饲等特性[2]。长白猪作为西方猪种,具有生长速度快、饲料利用率高、瘦肉率高等特点,但肉质欠佳[3]。长白猪是典型的瘦肉型猪,而松辽黑猪是我国特有的脂肪型猪,2个不同品种猪的表型差异很大,是鉴别肌肉和脂肪中DEGs的良好动物模型。
高通量测序使转录组研究变得容易进行,以转录组测序(RNA-seq)为代表的新一代高通量测序技术能够全面快速获取某一物种特定器官或组织在某一状态下的几乎所有转录本信息。该技术既可以在有基因组注释的情况下应用以促进转录本识别,也可以在没有参考基因组的情况下应用,其被广泛应用于转录组分析。王志秀[4]对滇南小耳猪、藏猪、长白猪和杜洛克猪进行高通量测序发现,在滇南小耳猪-藏猪组和长白猪-杜洛克猪组共筛选出315个差异表达基因,其中,有140个上调基因,175个下调基因,并鉴定出27个与脂肪沉积相关的基因,其主要参与脂肪代谢、脂肪酸合成等过程。Xu等[5]对不同日龄梅山猪的骨骼肌进行转录组研究,鉴定出338个DEGs,蛋白质谱分析发现66个差异表达基因,功能分析显示其功能主要为代谢、肌原纤维细丝形成、细胞骨架形成、收缩活动和信号转导。NOISeq是用于分析RNA-seq数据的综合资源,是一种用于鉴别DEGs的非参数方法,不需要对数据进行分布假设,就可以对原始计数或之前归一化或转换的数据集进行差异表达分析。由于噪声分布来自实际数据,NOISeq方法能更好地适应数据集的大小,可更有效地控制错误发现率[6]。
背最长肌是研究猪肉品质最常用的材料。本研究选取了6头松辽黑猪与6头长白猪,利用其背最长肌组织的转录本进行RNA-seq分析,通过非参数检验法NOISeq鉴定差异表达基因,并对差异表达基因的功能进行注释分析,以筛选出影响猪脂肪沉积的关键基因。
1 材料和方法
1.1 试验动物
本研究选用来自天津市宁河原种猪场的6头松辽黑猪与6头长白猪,所有动物在同一条件下饲养至体质量为100 kg左右时进行屠宰,屠宰在华都阳光食品公司进行,屠宰标准按照GB/T 17236—2008《生猪屠宰操作规程》进行[7]。屠宰后,采集背最长肌组织样品,并移至液氮中进行冷冻保存,待提取RNA使用。
1.2 总RNA的提取、建库及测序
采用TRIzol法,按照产品说明提取每头猪背最长肌组织的总RNA,共提取12个样本,然后用1%琼脂糖凝胶电泳初步检测总RNA是否有降解及杂质,并用紫外分光光度计测定波长,评价所得RNA质量。cDNA文库制备与测序在广州基迪奥生物科技有限公司进行。参考Illumina Tru Seq TM RNA样品制备试剂盒操作说明进行构建,最后用构建好的文库通过Illumina HiSeq 2500平台进行双末端(Paire-end,PE)测序。
1.3 测序数据的处理
利用Trimomatic[8]对原始数据(raw reads)进行过滤,并利用FastQC软件[9](http://www.bioinformatics.babraham.ac.uk/projects/fastqc/)对raw reads进行质控并统计。质控标准为:去除含 adapter 的 reads;去除含N比例大于10%的reads;去除低质量reads(质量值Q≤20的碱基数占整条read的50%以上)[10]。质控后获得高质量的序列数据(Clean reads),用TopHat2[11]将其比对到猪的参考基因组Susscrofa 11.1中。Cufflinks[12]用于数据的转录本组装、注释与合并。用HT-seq软件[13]计算每个样本中基因的表达量。
1.4 差异表达基因的筛选
用NOISeq R数据包筛选差异表达基因,以log2|fold change|>1、P≥0.8为标准进行筛选。NOISeq统计方法是基于无参估计鉴定差异表达基因的,所以并没有常见的P值,该方法的优点在于能够根据测序数据的大小对假阳性率进行有效控制,所采用的阈值P=0.8意味着该基因有差异表达的可能性是无差异表达的4倍,与其他方法中(cufflink、DESeq、DEGSeq等)所用的P=0.001筛选标准相持平[14]。
1.5 基因的生物学功能分析
GO(http://www.geneontology.org)为基因数据提供功能分类,包括生物过程(Biological process,BP)、分子功能(Molecular function,MF)和细胞成分(Cellular component,CC)3个方面。GO分析是一种广泛使用的基因和基因产物注释工具。通过GO term注释的差异基因,计算每个term的基因列表和基因数目,应用超几何检验,找出与整个基因组背景相比差异表达基因中显著富集的GO条目,从而找出差异表达基因显著相关的生物学功能。KEGG(http://www.genome.ad.jp/kegg/)是一个网络网站,可以分析、解释和可视化基因功能,是通路分析的主要数据库[15]。本研究利用基迪奥生物信息云平台(https://www.omicshare.com/)对差异表达基因进行GO功能富集分析和KEGG通路富集分析,P<0.05时被鉴定为显著富集通路和GO条目。
2 结果与分析
2.1 测序数据的质控及比对信息
对每个样本的测序数据质控后获得约1.0×107个Clean reads,与猪的参考基因组序列(Sus scrofa 11.1)比对,平均比对率约为88.8%。通过对转录本进行组装和拼接,最终得到23 471个转录本(FPKM≥0.1)。由测序样本的reads数和比对率可知,样本质量和测序测量均达到了预期(表1)。

表1 样本比对结果统计Tab.1 Statistics of sample comparison results
2.2 基因表达整体分析
为了了解reads在不同样本中的分布,比较了试验中所有样本中的reads数,发现基因的表达水平基本相当(图1-A),只有不到35%的基因在每一个样本中都有1个以上的CPM(图1-B)。

A为动态表达范围图,该图显示了每个样本中CPM值超过0的基因的分布;B图反映了低表达基因在总转录本中所占的比例。A is the dynamic expression range map,which shows the distribution of differentially expressed genes with CPM values over 0 in each sample;B map reflects the proportion of low-expressed genes in total transcripts.
2.3 生物型检测
分别选取松辽黑猪和长白猪中的1个样本进行生物型检测,发现检测到的大多数转录本为蛋白质编码RNA,有少部分RNA也能被检测到,如miRNA、lincRNA和scaRNA等,检测结果符合预期(图2)。

图2 生物型分布Fig.2 Biotype distribution(per sample)
2.4 差异表达基因筛选
在松辽黑猪和长白猪的背最长肌组织样本中共检测到23 471个基因表达(FPKM≥0.1)。2个不同品种猪相比,共筛选到664个差异表达基因(图3),其中,364个基因在松辽黑猪中高表达,300个基因在长白猪中高表达。

A图代表2个品种猪的表达值总图,其中,红点代表差异表达基因;横坐标代表基因在长白猪中的表达水平,纵坐标代表基因在松辽黑猪中的表达水平。B图代表(M,D)值和差异表达基因汇总图。横坐标表示值,而纵坐标表示值,黑点表示(M,D)值,其中(|M|>1),红点表示差异表达基因。
2.5 差异表达基因的功能富集分析
为了更好地了解所筛选到的DEGs的功能及其与表型的关系,本研究对筛选到的DEGs进行了功能富集分析,包括GO功能分析和KEGG信号通路分析。GO功能富集分析发现,差异表达基因富集于52条GO条目,且生物过程富集到的GO terms最多,由此可知,生物过程是松辽黑猪和长白猪在背最长肌中差异表达基因行使的主要生物学功能(图4)。对DEGs进行KEGG信号通路富集分析,发现这些基因共参与了275条KEGG信号通路,其中,有52条显著富集通路,其通路主要为脂肪酸代谢(Fatty acid metabolism)、胰岛素抵抗(Insulin resistance)、PPAR信号通路(PPAR signaling pathway)、AMPK信号通路(AMPK signaling pathway)、脂肪细胞因子信号通路(Adipocytokine signaling pathway)和胰岛素信号通路(Insulin signaling pathway)等(图5),根据显著性通路筛选到LPIN1、FADS1、FADS2、PLIN2、PPARGC1A、PRKAG2和ACSL1等与脂类代谢和肌肉发育相关的基因(表2)。
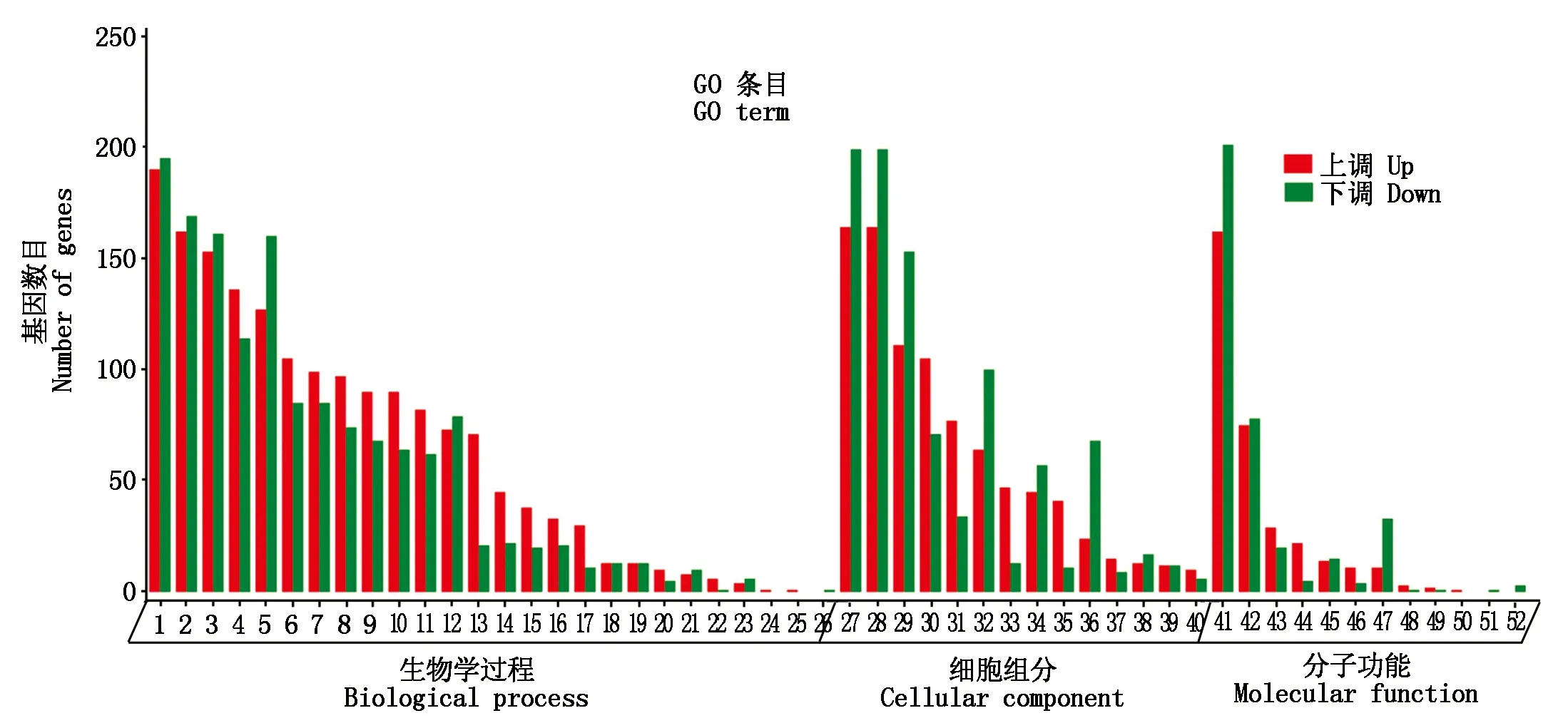
1.细胞过程; 2.生物调节; 3.生物过程的调节; 4.对刺激的反应; 5.代谢过程; 6.多细胞有机体过程; 7.生物过程的正向调控; 8.信令; 9.发展过程; 10.定位; 11.细胞成分组织或生物发生; 12.生物过程负调控; 13.免疫系统过程; 14.运动; 15.多组织进程; 16.细胞增殖; 17.生物黏附; 18.繁殖; 19.生殖过程; 20.行为; 21.成长; 22.细胞杀伤; 23.有节奏的过程; 24.解毒; 25.色素沉着; 26.细胞聚集; 27.细胞; 28.细胞部分; 29.细胞器; 30.膜; 31.膜部分; 32.细胞器部分; 33.细胞外区; 34.含蛋白质的复合物; 35.胞外区部分; 36.膜封闭内腔; 37.突触; 38.超分子配合物; 39.细胞连接; 40.突触部分; 41.黏合物; 42.催化活性; 43.分子功能调节剂; 44.分子换能器活性; 45.运输活动; 46.结构分子活性; 47.转录调节活性; 48.抗氧化活性; 49.自噬受体活性; 50.蛋白质标签; 51.分子载体活性; 52.翻译调节活动。
3 结论与讨论
本研究选用在表型方面存在较大差异的松辽黑猪和长白猪为研究对象,对其背最长肌组织进行高通量转录组测序,筛选与脂肪沉积相关的差异表达基因。通过非参数检验法NOISeq共筛选出664个DEGs,其中,364个基因在松辽黑猪中高表达,300个基因在长白猪中高表达。对其进行生物学功能分析,鉴定出脂肪细胞因子信号通路、PPAR信号通路、AMPK信号通路、胰岛素信号通路和脂肪酸代谢等与脂肪沉积相关的通路,根据显著性通路筛选出了LPIN1、FADS1、FADS2、PLIN2、PPARGC1A、PRKAG2和ACSL1等与脂类代谢和肌肉发育相关的基因。该研究结果为深入研究松辽黑猪和长白猪之间脂肪沉积和肌肉发育性状差异的分子机制提供了坚实的基础。猪脂肪组织的沉积能力具有品种差异,且不同品种的猪脂肪沉积能力具有较大差异[16]。本研究找到了与松辽黑猪和长白猪脂肪沉积相关的重要功能基因和信号通路,如mTOR信号通路(mTOR signaling pathway)、PPAR信号通路和脂肪酸代谢,与之相关的基因包括脂质1基因(Lipin 1,LPIN1)、脂滴蛋白2基因(Perilipin-2,PLIN2)和脂肪酸去饱和酶1基

图5 KEGG通路富集分析Fig.5 KEGG pathway enrichment analysis
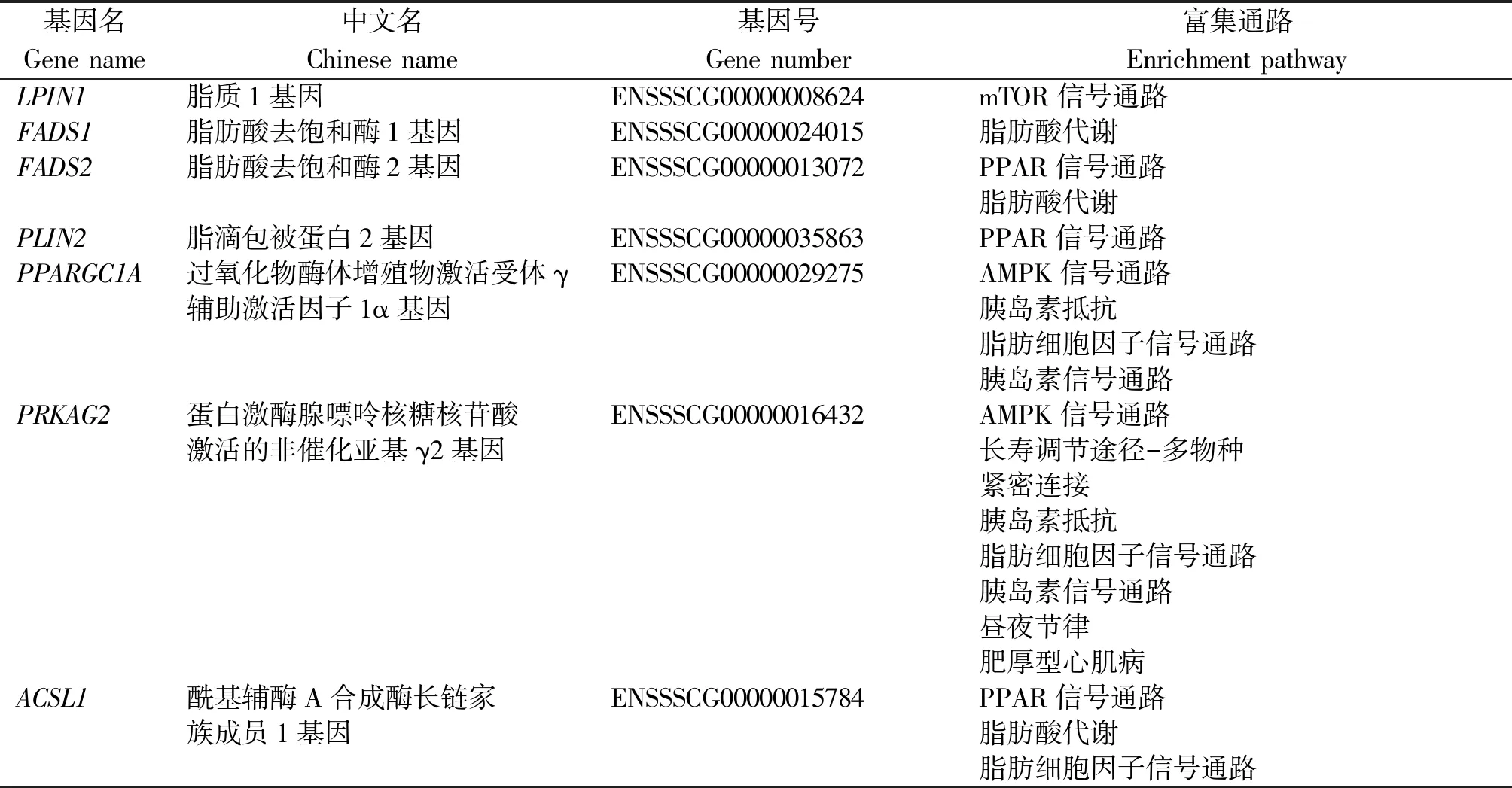
表2 脂质代谢相关KEGG通路Tab.2 Lipid metabolism-related KEGG pathway
因(Fatty acid desaturase 1,FADS1)。LPIN1基因的表达是脂肪细胞分化所必需的,其作为核转录共激活因子与一些过氧化物酶体增殖物激活受体一起调节参与脂质代谢其他基因的表达。He等[17]研究发现,在猪中LPIN1基因与肌内脂肪沉积及肌肉品质存在显著相关性,可以影响肌内脂肪含量(IMF)、瘦肉率和风味,所以将LPIN1基因作为改善肌肉品质的候选基因之一。PLIN2基因位于脂滴表面,称为脂滴包被蛋白,隶属于脂滴包被蛋白PAT家族蛋白,其主要功能是参与脂质代谢[18]。有研究表明,PLIN2基因在肌间脂肪较高的猪种中表达水平较高[19]。FADS1基因是脂肪酸去饱和酶(FADS)基因家族的成员,去饱和酶通过在脂肪酰基链的定义碳之间引入双键来调节脂肪酸的不饱和度。宋文莉等[20]研究发现,FADS1基因对ω-3不饱和长链脂肪酸的代谢有重要作用,加之FADS1基因在本研究中参加了脂肪酸代谢,因此,推测FADS1基因为与猪脂肪沉积相关的候选基因,目前关于FADS1基因在猪上的研究还很少。另外,发现FADS2、过氧化物酶体增殖物激活受体γ辅助激活因子1α基因(Peroxisome proliferators activated receptor γ coactivator1α,PPARGC1A)、蛋白激酶腺嘌呤核糖核苷酸激活的非催化亚基γ2基因(Protein kinase AMP-activated non-catalytic subunit gamma 2,PRKAG2)和长链脂酰辅酶A合成酶1基因(Long chain acyl-CoA synthetase 1,ACSL1)均同时参与了不同信号通路和代谢途径。FADS2属于脂肪酸去饱和酶(FADS)基因家族的成员,主要作用为通过在脂肪酰基链的特定碳原子之间引入双键来调节脂肪酸的不饱和度。本研究发现,FADS2基因在PPAR信号通路和脂肪酸代谢通路中均有参与,前人研究发现,FADS2基因与多不饱和脂肪酸(PUFA)之间存在关联[21]。另有研究发现,FADS2基因SNP突变影响人体脂肪组织及血浆中PUFA的含量,从而影响人类健康,人类全基因组关联分析证实,FADS2基因对脂类代谢疾病有重要作用[22];因此,推测FADS2基因可能在猪脂肪沉积中也发挥着重要作用。前人研究发现,PPARGC1A的表达有利于脂肪细胞的分化成熟,与脂质代谢相关[23],且PPARGC1A基因对生物体内棕色脂肪的生成具有重大影响[24]。Gandolfi等[25]研究发现,PPARGC1A基因与猪肉质性状存在显著关联,且由于物种间存在差异,可作为候选基因用于猪肉质性状的改良。本研究中,PPARGC1A基因参与了脂肪细胞因子信号通路、AMPK信号通路和胰岛素抵抗等多条通路,所以,可将PPARGC1A基因作为猪背脂沉积的重要候选基因之一。PRKAG2参与了AMPK信号通路、脂肪细胞因子信号通路和胰岛素信号通路等多条通路,PRKAG2基因是腺嘌呤核糖核苷酸活化蛋白激酶(AMP-activated protein kinase,AMPK)家族重要的成员,其突变与Wolff-Parkinson-White综合征、家族性肥厚性心肌病和心脏糖原贮积病有关。其在肌肉脂肪代谢、能量代谢等方面发挥着重要作用[26]。Jing等[27]通过对高、低剩余采食量的猪骨骼肌进行高通量测序发现,PRKAG2在骨骼肌能量代谢过程中起着至关重要的作用。由此推测,PRKAG2基因的表达与猪的脂肪沉积存在显著相关性,可作为影响脂肪沉积的重要候选基因。ACSL1是长链酯酰辅酶A合成酶家族(Long-chain fatty acyl-CoA synthetases,ACSLs)的成员之一,该家族由ACSL1、ACSL3、ACSL4、ACSL5和ACSL6组成[28],而ACSL1是ACSLs家族中最早被发现的亚型,ACSL1在脂质生物合成和脂肪酸降解中发挥关键作用。有研究表明,ACSL1在脂肪酸的活化、转运和降解以及脂类生成等过程中发挥着至关重要的作用[29]。本研究中,ACSL1基因参与了PPAR信号通路、脂肪酸代谢和脂肪细胞因子信号通路,数量性状基因座(Quantitative trait loci,QTL)分析认为,ACSL1作为最重要位置和功能候选基因,影响猪肉中脂肪酸组成[30];因此,将ACSL1基因作为脂肪沉积的候选基因。
综上所述,通过对松辽黑猪和长白猪背最长肌之间差异表达基因进行鉴定与分析,筛选出多个参与肌肉发育和脂类代谢的重要候选基因,包括LPIN1、FADS1、FADS2、PLIN2、PPARGC1A、PRKAG2和ACSL1等,这些基因目前已知的功能均与脂肪沉积相关,但在猪中影响组织脂类代谢的机制还有待深入研究,未来对这些重要功能基因的深入验证,可能会揭示影响猪生长发育和猪肉品质的重要基因,为未来满足社会需求的新品种遗传育种提供理论基础。
猜你喜欢
当代畜禽养殖业(2023年5期)2023-11-20 12:50:56
今日农业(2022年7期)2022-07-06 20:28:39
江苏农业科学(2021年18期)2021-09-28 23:15:44
金秋(2020年10期)2020-08-22 13:11:50
读友·少年文学(清雅版)(2019年1期)2019-05-09 02:35:26
中国稻米(2018年2期)2018-04-18 12:37:17
华南农业大学学报(2015年5期)2015-12-04 03:04:38
娃娃画报(2014年9期)2014-10-15 16:31:59
小朋友·快乐手工(2014年1期)2014-02-25 12:19:55
