
超临界CO2辅助聚己二酸对苯二甲酸丁二醇酯/聚乳酸珠粒发泡
2022-11-02 12:21:16谢茂卿杜俊威张世豪胡园园刘清亭付旭东胡圣飞
高分子材料科学与工程 2022年8期
谢茂卿,杜俊威,张世豪,胡园园,刘清亭,付旭东,张 荣,胡圣飞
(1.湖北工业大学绿色轻工材料湖北省重点实验室;2. 湖北工业大学材料与化学工程学院,湖北 武汉 430068)
发泡材料是一种功能性优异的新型材料,由于其介电常数低、绝缘隔热、吸音降噪、质量轻等突出优点被广泛应用于绿色包装、医学等领域[1,2]。聚己二酸对苯二甲酸丁二醇酯(PBAT)具有较高的断裂伸长率、优异的韧性、良好的加工性能[3]和全降解特性,近年来被广泛应用于发泡领域。但PBAT 存在低模量,低熔点,低结晶度[4]等缺陷,使得PBAT 在发泡过程中,其分子链被拉伸,结晶度低而无法在室温定型[5]。其发泡珠粒存在收缩、合并及破裂等现象,泡孔密度及尺寸调控受到限制。
低结晶度及分子链中存在大量柔性链段的泡沫材料,其自身熔体强度不足,泡孔内高浓度二氧化碳(CO2)极易向空气扩散,在内部形成负压,导致泡沫收缩及泡孔塌陷[6]。有学者为改善热塑性聚氨酯(TPU)和低密度聚乙烯(LDPE)泡沫的收缩性,采用一步泄压法将CO2与氮气(N2)混合分压,利用N2的稳定性和CO2的高渗透率,减缓泡孔内部CO2的扩散速度,改善泡沫的收缩性[7~9],但N2的引入在一定程度上会降低其发泡倍率。Wang 等[10]则采用一步泄压法以热空气作介质,将聚乳酸(PLA)与TPU 共混来提高TPU 的熔体强度,利用PLA 分子链和晶体限制TPU 分子链运动,抑制TPU 泡沫的收缩,将收缩率降低至29%。而Zhang 等[11]采用一步泄压法以水作介质,将TPU 与马来酸酐(MAH)接枝后与丙烯腈-丁二烯-苯乙烯(ABS)共混,提高TPU/ABS 的熔体强度,以此抑制泡孔收缩。因此,共混改性将是改善发泡性能简单有效的方法。为不损失PBAT 的可降解性能,将高熔点的PLA(175 ℃)与低熔点的PBAT(125 ℃)共混以此提高PBAT 的熔体强度,改善其发泡性能和抗收缩性能。
一步泄压法制备的发泡珠粒在不添加介质时珠粒表面易黏结,而采用水作为介质时,PBAT 和PLA又极易水解。因此本文采用升温法发泡有效解决了珠粒黏结问题,制备了一种抗收缩、表面光滑不黏结的PBAT/PLA 珠粒发泡材料;讨论了以高熔点PLA 作为网络骨架来改善PBAT/PLA 的发泡性能及发泡珠粒的抗收缩性;进一步研究了结晶性能和流变性能对共混物发泡性能和收缩性能的影响。
1 实验部分
1.1 主要原料
聚己二酸对苯二甲酸丁二醇酯:TH801T,密度为1.25 g/cm3,熔融指数5 g/10 min,新疆蓝山屯河科技股份有限公司;聚乳酸:2003D,密度1.24 g/cm3,熔融指数20 g/10 min,美国Nature Works 公司;CO2:纯度99.5%,武汉市博强气体有限公司。
1.2 制备步骤和工艺
1.2.1 PBAT/PLA 共混物颗粒的制备:先将PBAT 和PLA 颗粒置于真空干燥箱中干燥,再将不同质量比的PBAT/PLA(100/0,99/1,97/3,95/5,93/7,91/9)颗粒混合均匀,通过单螺杆挤出机(150 ℃,165 ℃,180 ℃,170 ℃)制得共混物小颗粒。
1.2.2 PBAT/PLA 共混物发泡珠粒的制备:将干燥后不同PLA 含量的共混物颗粒置入高压釜中,通入CO2气体对高压釜吹扫并排空30 s,开始升温至50 ℃,后通入CO2到14 MPa,保压3 h,泄压转移颗粒置入130 ℃的恒温箱中发泡40 s 得到共混物发泡珠粒。
1.3 测试与表征
1.3.1 PBAT/PLA 共混物结晶性能分析:利用差示扫描量热仪(DSC, 8500 型,美国PE 公司)测定共混物的热力学行为。将螺杆挤出的PBAT/PLA 共混物小颗粒分为2 组,一组置于50 ℃的高压釜中,不通入CO2气体,维持3 h;另一组置于50 ℃的高压釜中,通入CO2气体至14 MPa,维持3 h。取出2 组样品立即进行DSC 测试。在氮气氛围下,设定升温/降温速率为10 ℃/min,温度范围为30~200 ℃。采用加热、冷却模式。测试2 组样品DSC 的第1 条升温曲线和降温曲线。并利用DSC 升温熔融曲线计算PBAT 的相对结晶度,计算公式如式(1)
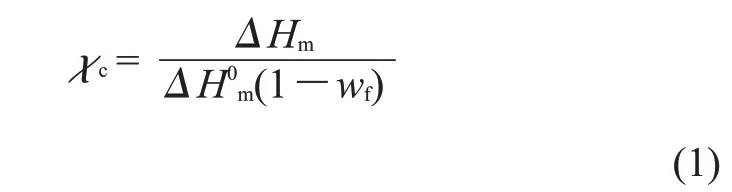
式中:χc——结晶度;ΔHm——PBAT 的实际熔融热焓;ΔH0m——PBAT 的100%熔融热焓,其值为114 J/g;wf——为共混物中PLA 的质量比。
利用X 射线衍射仪(XRD, D8 Advance 德国Bruker 公司)测定共混物在室温下的结晶形态,扫描角度(2θ)为5°~70 °,速率为5 (°)/min。
利用偏光显微镜(POM, DM 4500P, 德国Leica公司)测定共混物的晶体形态。取约2 mg 共混物样品置于热台载玻片上,升温/降温速率为10 ℃/min。升温30~200 ℃时在200 ℃压制成约50m 厚的薄膜并维持2 min 消除热历史,后降温至50 ℃。一组维持3 h 观察晶体形态,再升温至130 ℃维持40 s 观察晶体形态;另一组取出薄片置于50 ℃的高压釜中,通入CO2压力至14 MPa,维持3 h 后取出,观察此时的晶体形态。
1.3.2 PBAT/PLA 共混物流变性能测试:利用旋转流变仪(MCR92, 奥地利Anton Paar 公司)测定共混物在130 ℃的流变性能。采用直径为25 mm、间隙为1 mm 的平行板进行动态振动扫描,频率范围为0.1~100 rad/s,应变设置为1%。
1.3.3 PBAT/PLA 发泡性能测试:采用扫描电子显微镜(SEM,JSM-6390LV,日本JEOL 公司)观察发泡珠粒的断面形态。将发泡样品置入液氮冷冻脆断,取其断面进行喷金处理后观察分析。使用Image pro 软件计算SEM 图片得到泡孔直径和泡孔密度,式(2)、式(3)为其计算公式[12]
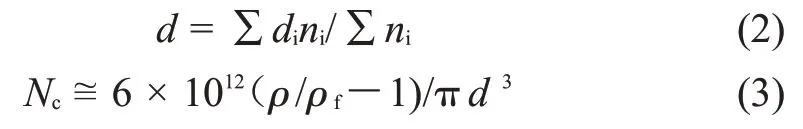
式中:d——数均泡孔直径m;ni——当量直径,di——泡孔数;Nc——泡孔密度,cm-3;ρƒ和ρ——分别为发泡样品密度和未发泡样品密度,g/cm3。
1.3.4 PBAT/PLA 收缩性测试:度量PBAT/PLA 发泡珠粒的收缩性通过测量发泡珠粒的密度计算前后的体积膨胀率(φ1,φ2),发泡珠粒的收缩率(S)用式(4)计算
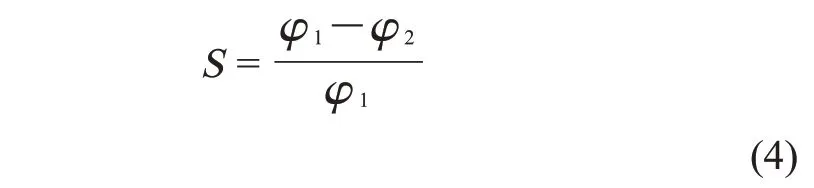
式中:φ1——初始发泡倍率;φ2——最终发泡倍率。φ1——发泡结束后立即测量得的发泡珠粒的发泡倍率;φ2——发泡珠粒放置48 h 后测量的发泡珠粒的发泡倍率。
2 结果与讨论
2.1 PBAT/PLA 共混物的流变性能
聚合物的流变性能对其发泡性能的影响极其明显。Fig.1 为不同PLA 含量时PBAT/PLA 共混物的储能模量(G′)、损耗模量(G′)、复数黏度(η*)及Cole-Cole 曲线随频率变化的关系图。从Fig.1(a)和Fig.1(b)可知,在低频区,PBAT/PLA 共混物的G′随着PLA 含量的增加而增加,且均高于纯PBAT,G′表现出相同的趋势,表明共混物的黏弹性增强。这是因为PLA 刚性链段的引入使得PLA 与PBAT 的分子链之间形成更多的物理交联点,增加了共混物的刚性。Fig.1(c)为PBAT/PLA 共混物的η*随频率变化的关系曲线。由图可知,共混物的η*均随频率的增加而减小,呈现出典型的剪切稀化行为。在低频区,随着PLA 含量的增加,共混物的η*增大,这是因为PBAT 与PLA 的分子链之间产生了物理交联和链缠结,在低频剪切过程中难以解缠,使η*增加,从而有利于改善泡孔形态及提高泡孔的抗收缩性能。当PLA 质量分数为5%时,共混物具有一定熔体强度,在泡孔生长过程中利于泡孔的稳定,并减少泡孔破裂、合并等情况发生。当PLA 质量分数超过5%时,共混物黏度过高将限制泡孔生长,不利于发泡。Fig.1(d)为共混物的Cole-Cole 曲线。结果表明,纯PBAT 的Cole-Cole 曲线接近半圆状,而PBAT/PLA 共混体系的Cole-Cole 曲线明显偏离了半圆弧形状,其末端出现上扬,且随着PLA 含量的增加,曲线偏离半圆状程度越高,末端上扬程度更大,表明共混物中存在松弛时间更长的链[13],而这些松弛时间更长的链是共混物中PBAT 与PLA 分子链的缠结形成的。
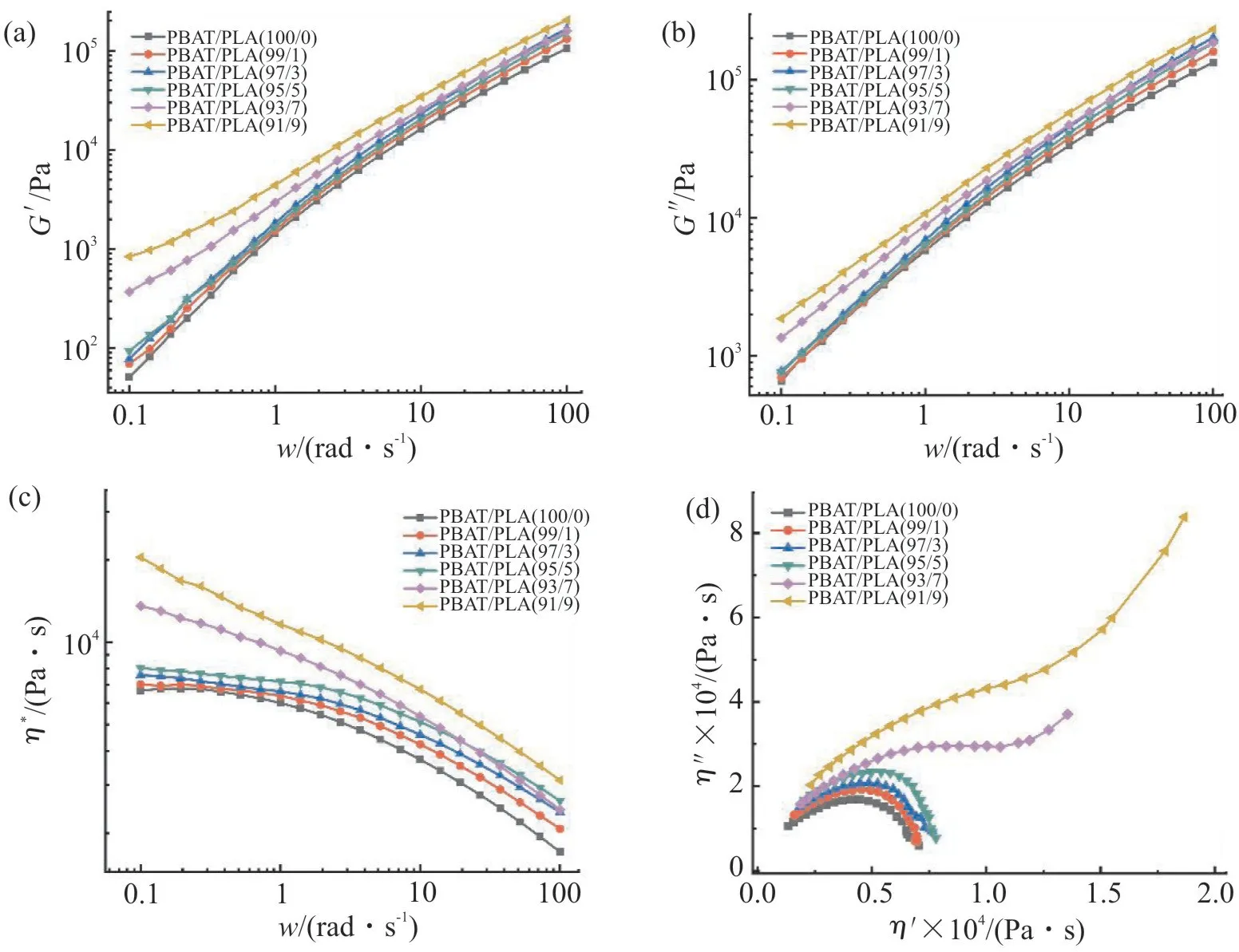
Fig.1 Rheological properties of PBAT/PLA blends
2.2 PBAT/PLA 共混物结晶性能
Fig.2 是不同PLA 含量的PBAT/PLA 升温和降温结晶DSC 曲线及经scCO2浸渍(50 ℃,14 MPa,3 h)后的DSC 曲线,通过对熔融热焓(ΔHm)及结晶度(χc)的计算得到Tab.1。从Fig.2(a)中PBAT/PLA 共混物的升温曲线可知,PLA 的熔融温度(Tm)为175 ℃、PBAT 的Tm为122 ℃。随 着PLA 含 量 的 增加,PBAT 的熔融峰略向高温方向偏移,而PLA 的熔融峰几乎不发生偏移,表明PBAT 与PLA 的相容性较弱[14]。Fig.2(b)为共混物经过scCO2浸渍后的升温结晶曲线。结果表明,与未经scCO2浸渍的相比,浸渍后共混物中PBAT 的ΔHm 和χc都增加,这是由于scCO2流体的作用促进了PBAT 的分子链运动,有利于链段取向,使结晶度增加。而PLA 的熔融峰面积减小,表明PLA 的结晶度减弱,这是因为scCO2促进了两相互容[15],PBAT 与PLA 分子链缠结更加紧密,PBAT 分散于PLA 中破坏了PLA 的有序性使之更难形成晶体,减弱了PLA 的结晶。Fig.2(c~d)为共混物经scCO2浸渍前后的结晶温度,由图可知,未经过scCO2浸渍的共混物的结晶温度约为72 ℃,而浸渍后的共混物结晶温度提高到76 ℃。共混物的结晶性能增强,不仅增加了成核位点,促进了非均相成核,并且共混物晶区可以支撑泡孔,减少收缩,提高发泡倍率。
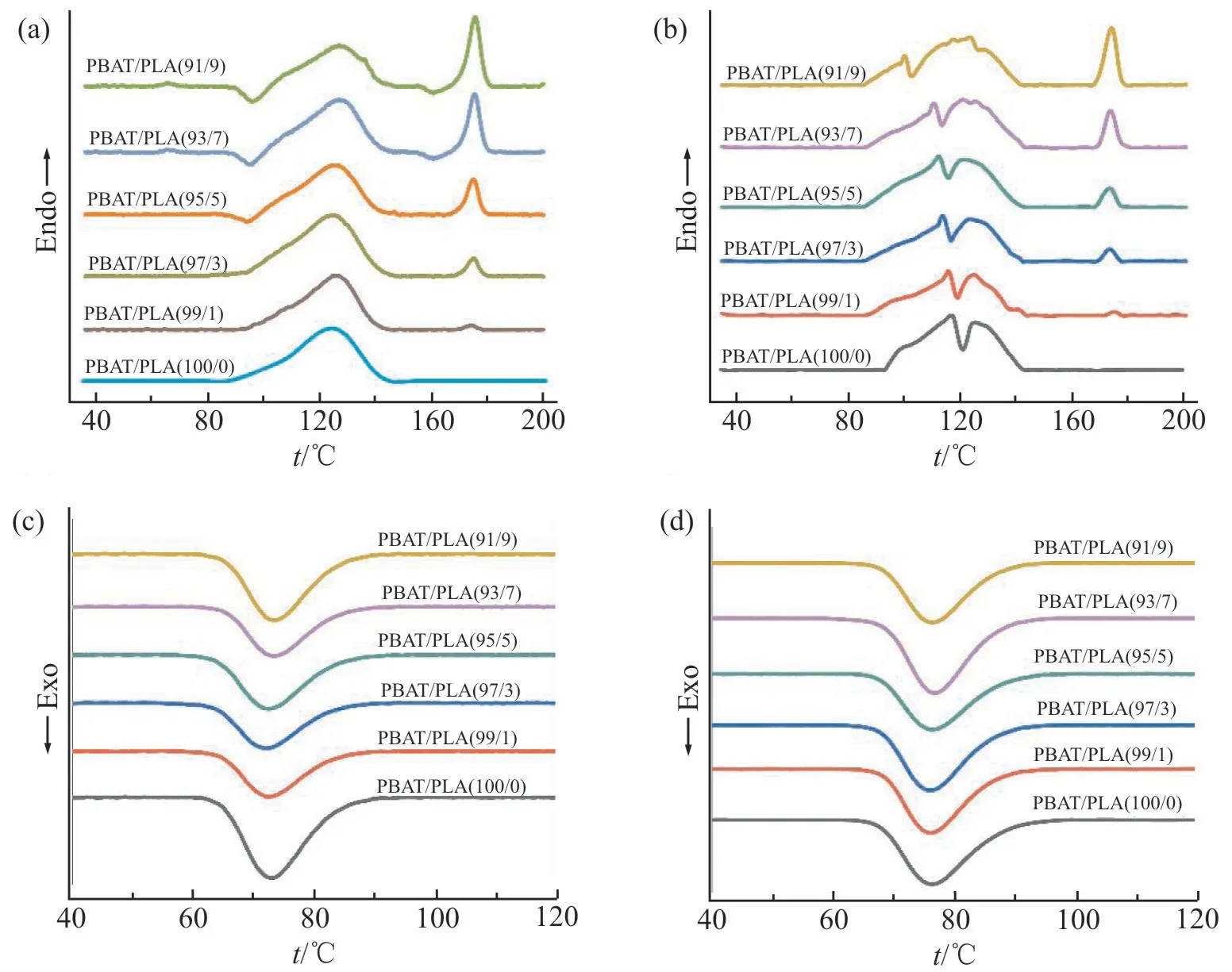
Fig.2 DSC curves of PBAT/PLA blends
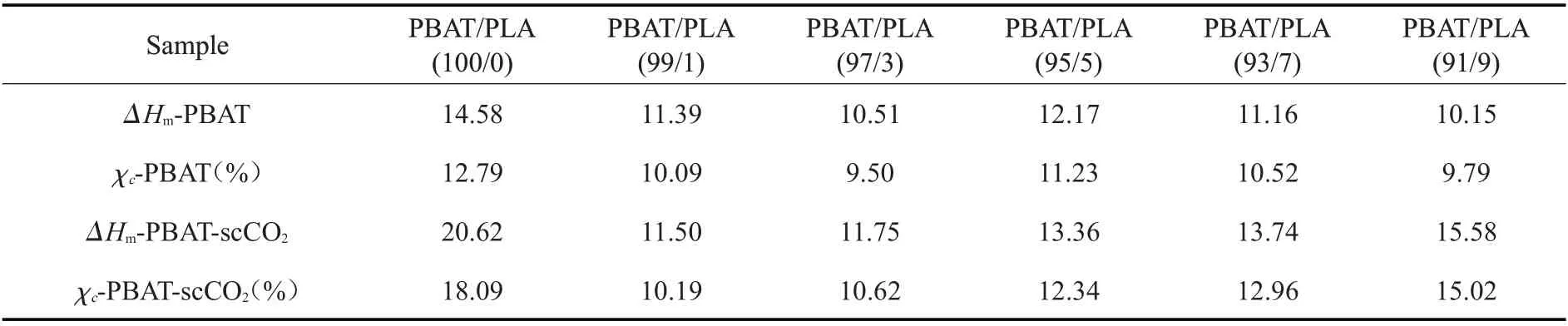
Tab.1 Melting enthalpy(ΔHm) and crystallinity(χ c) of PBAT component in PBAT/PLA blend before and after scCO2 impregnation
Fig.3 为PBAT/PLA 共混物经scCO2浸渍前后的XRD 曲线。Fig.3(a)为PBAT/PLA 共混物未浸渍的XRD 曲线,可以看出,纯PBAT 有5 个特征峰,分别为15.92°,17.26°,20.19°,22.94°和24.63°。纯PLA 的特征峰为16.64°和19.02°,随着PLA 含量的增加,PLA 峰位置不变、峰强度增加。Fig.3(b)是PLA 质量分数分别为0%,5%和9%的PBAT/PLA 共混物浸渍后的XRD 曲线。结果表明,浸渍后共混物中的PBAT 整体峰位置基本不发生偏移,但PLA 峰位置向左偏移,衍射峰强度降低,衍射峰范围变宽。根据Scherrer 公式[16],衍射峰范围变宽、晶体尺寸减小,表明PLA 的晶体尺寸变小。这是由于超临界流体的诱导作用,使PLA 中的部分α晶型转化为α′晶型[17],细化的晶体在发泡时能有力支撑泡孔结构。
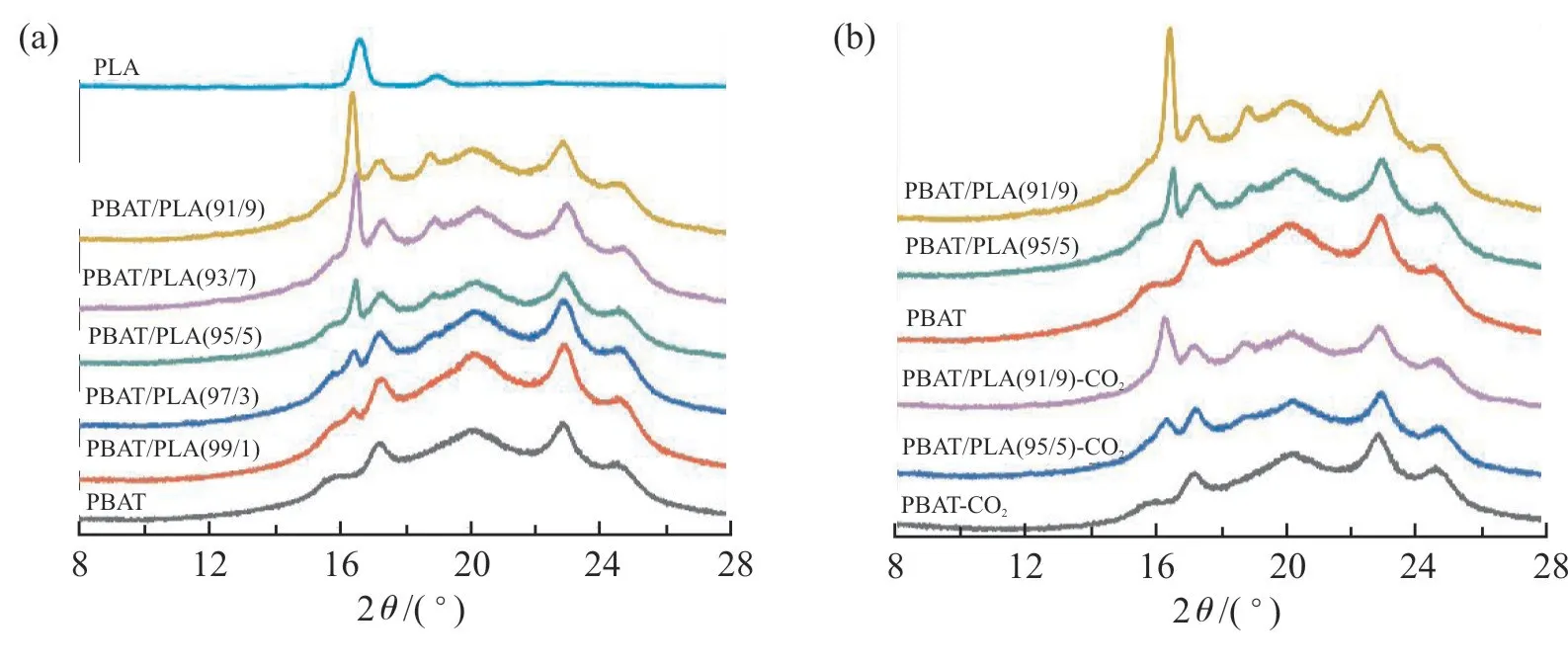
Fig.3 XRD patterns of PBAT/PLA blends
Fig.4 为PBAT/PLA 共混物在50 ℃时维持3 h 的晶体形态及由50 ℃升温至130 ℃的POM 照片。其中,Fig.4(a~g)和(a1~g1)分别为PBAT/PLA 共混物在50 ℃和130 ℃时的晶体形态,在50 ℃时,PBAT 的晶体是较小的球晶,PLA 晶体结构为不完善的球晶。随着PLA 含量增加,共混物中PLA 晶体数目增加,PBAT/PLA 共混物的晶体尺寸变小。当温度为130 ℃时,PBAT 晶体基本熔融,PLA 晶体此时不熔且进一步生长完善。Fig.4(h)为PLA 质量分数5%的PBAT/PLA 在50 ℃,14 MPa 压力浸渍后的晶体形态,经过scCO2的作用,晶体尺寸减小、晶体间隙增大、晶体数目增加、异相成核位点增加,将增加泡孔成核密度,有利于提高发泡倍率。

Fig.4 POM photos of PBAT/PLA blends for 5 μm
2.3 PBAT/PLA 发泡性能
Fig.5 为不同PLA 含量的PBAT/PLA 发泡珠粒的泡孔断面形态及泡孔分布,Fig.6 为不同PLA 含量下PBAT/PLA 发泡珠粒的平均泡孔直径、泡孔密度及体积膨胀率曲线图,与Fig.5 的泡孔结构一一对应。由Fig.5(a1)可以看出,纯PBAT 发泡珠粒的泡孔分布不均,孔壁较薄,泡孔破裂和并泡现象明显,平均泡孔尺寸较大,发泡倍率低至4.51 倍。由Fig.5(b1~d1)可观察到,当PLA 质量分数由1%增加到5%时,泡孔破裂的现象减少、泡孔尺寸显著降低、泡孔密度显著增加。PLA 质量分数达到5%时,此时的泡孔密度与体积膨胀率达到最大值,分别为7.49×107cm-3和9.47 倍。这是因为PLA 作为异质成核点,降低了泡孔成核的能垒、增加了泡孔成核的数量,泡孔尺寸减小,有利于均匀泡孔的产生[18]。当PLA 质量分数增加至9%时,如Fig.5(e1~f1)所示,泡孔壁较厚,未发泡区域增加。这是因为在发泡温度130 ℃时,未熔化的PLA 晶体在共混物中形成物理交联点,使PBAT/PLA 共混物的黏度和熔体强度增加,泡孔生长阻力增强,导致泡孔的平均尺寸减小。但共混物熔体黏度和弹性过高时,反而会限制泡孔的生长。Fig.5(a2~f2)显示了共混物的泡孔形态分布。由图可知,纯PBAT 的泡孔尺寸分布较宽且不均匀。随着PLA 含量的增加,PBAT/PLA 发泡珠粒的泡孔尺寸分布逐渐变窄。当PLA 质量分数为5%时,泡孔尺寸分布最符合正态分布,泡孔形态最优。

Fig.5 SEM photos of cell fracture surface and cell distribution of PBAT/PLA foamed beads
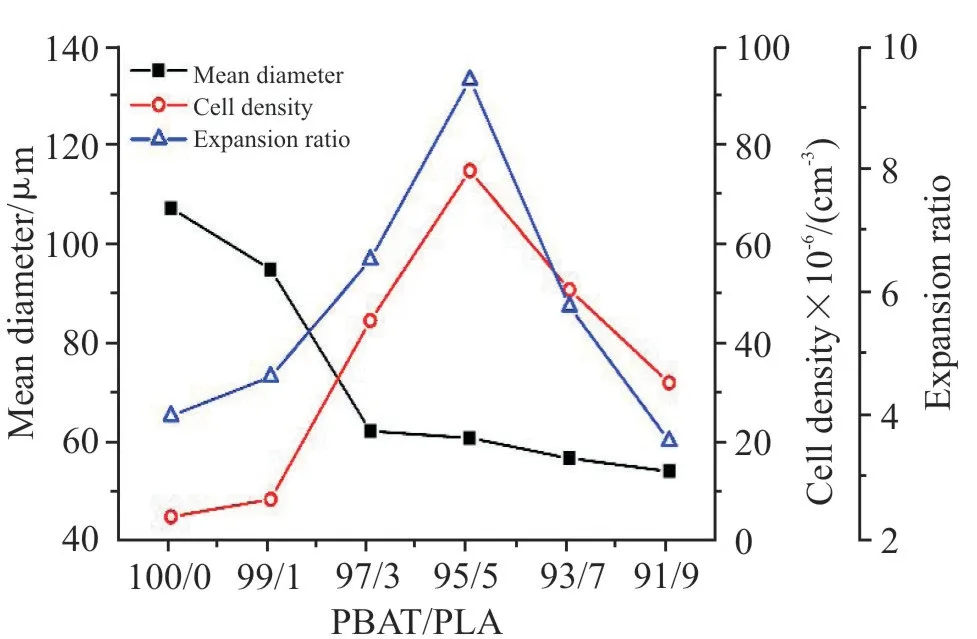
Fig.6 Cell density, mean cell diameter and expansion ratio of different foam beads
2.4 PBAT/PLA 发泡珠粒的收缩性
Fig.7 为不同PLA 含量时PBAT/PLA 发泡珠粒的收缩率。Fig.8(1)为PBAT/PLA 发泡珠粒0 h 原始实物图,Fig.8(2)为发泡珠粒空气中静置48 h 后收缩实物图。由Fig.7 可知,发泡珠粒的收缩率随着PLA含量的增加而降低,在PLA 质量分数超过5%时趋于稳定。当不添加PLA 时,纯PBAT 发泡珠粒的收缩率为24.1%,当PLA 质量分数增加至9%时,发泡珠粒收缩率降至5.6%。这是因为PBAT/PLA 黏度的增加使得泡孔壁强度增加,延长了泡孔内外CO2的扩散时间,且未熔的PLA 作为骨架结构,阻碍了分子链的恢复,降低了发泡珠粒的收缩性。从Fig.8(1)和Fig.8(2)中可以看出,原始发泡珠粒表面光滑,静置48 h 后,纯PBAT 发泡珠粒收缩最大,随着PLA 含量的增加,珠粒表面逐渐光滑,收缩性显著降低。
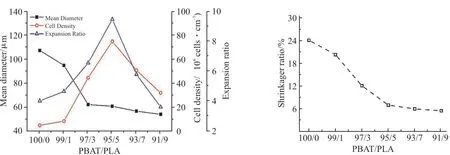
Fig.7 Shrinkage of PBAT/PLA foam beads with different PLA mass fractions
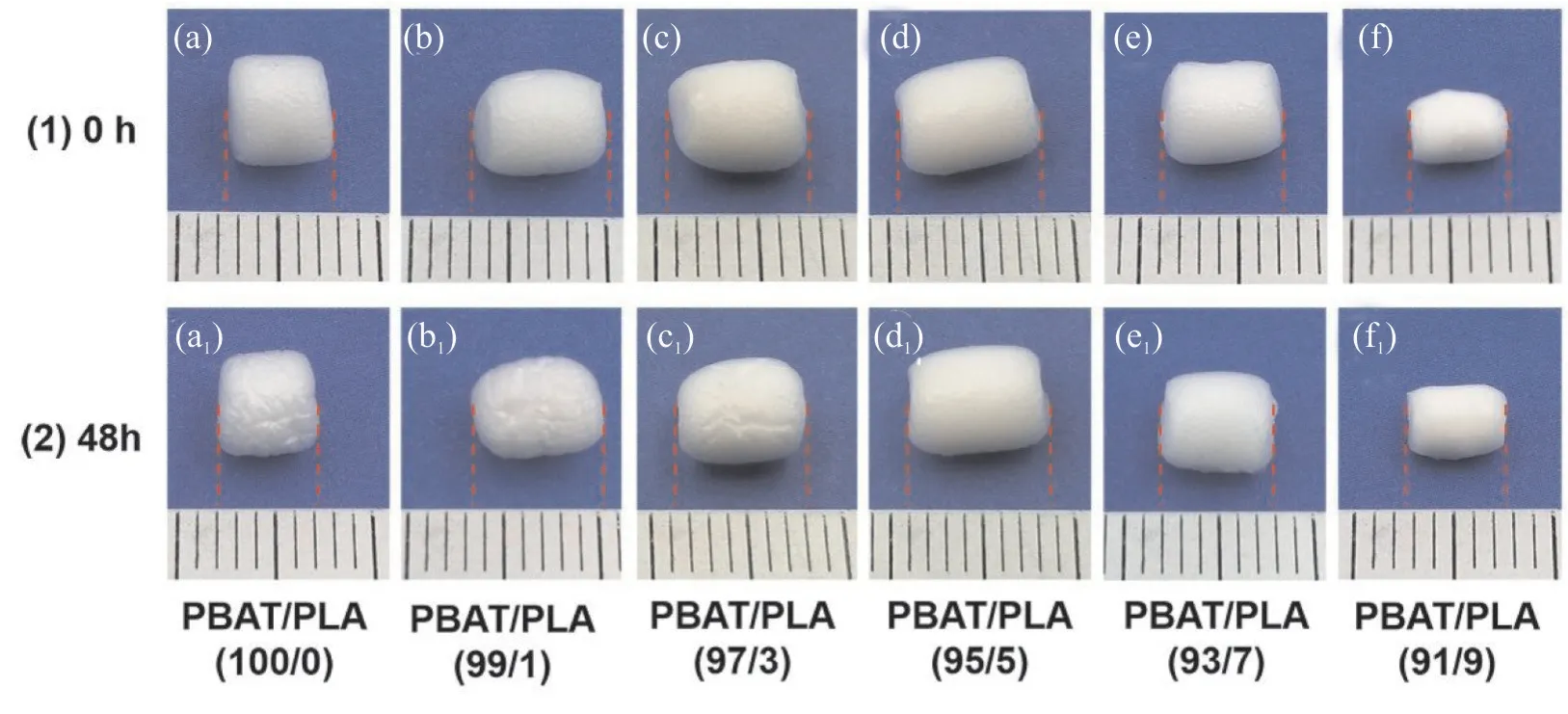
Fig.8 Shrinkage morphologies of PBAT/PLA foam beads
2.5 PBAT/PLA 发泡模型
Fig.9 为发泡过程中PBAT 与PLA 分子链变化模型 图。PBAT/PLA 经 过scCO2浸 渍 作 用 下,PBAT 的结晶性能得到一定增强。当在130 ℃迅速发泡时,由于发泡温度处于PBAT 与PLA 熔点之间,大部分PBAT 晶体熔化,此时PLA 高温晶体不熔,支撑住泡孔结构。当发泡完成时,PBAT/PLA 发泡珠粒此时在室温冷却,在冷却定型中再次结晶,此时熔体强度较发泡时增加,进一步支撑住泡孔稳定,从而延长了CO2的扩散时间,进而减少泡孔的收缩。
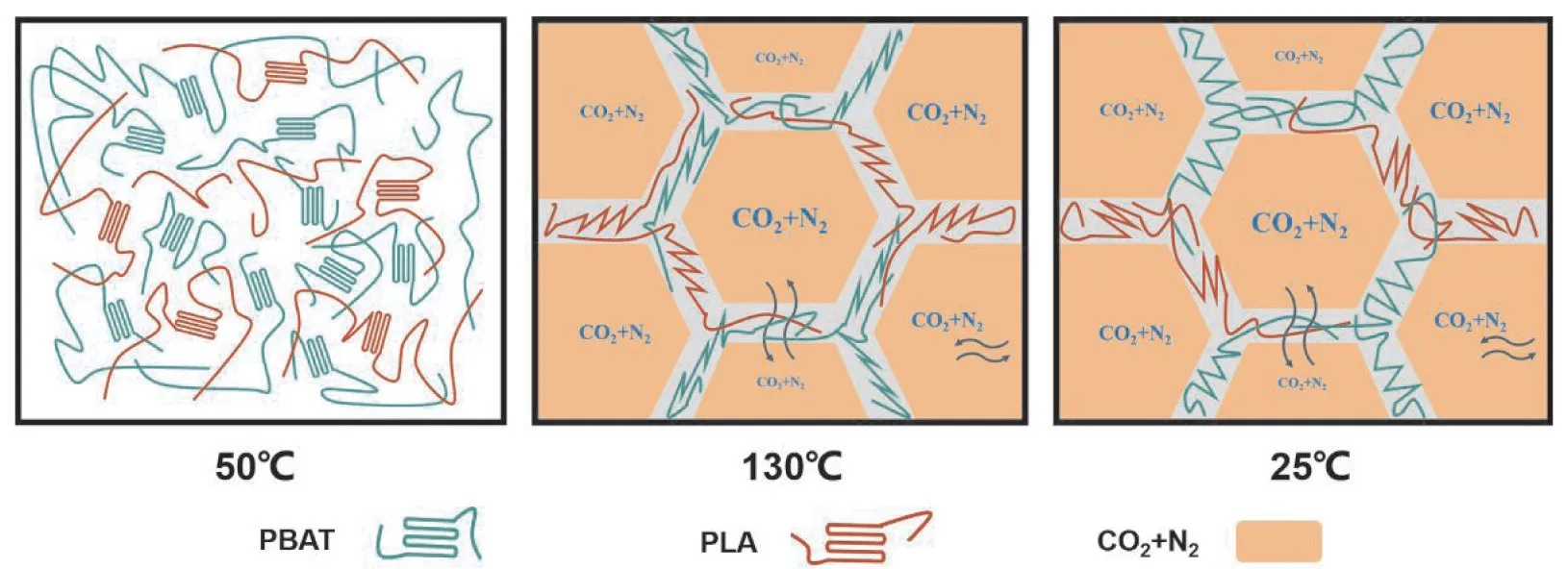
Fig.9 Model diagram of PBAT/PLA foam
3 结论
(1)在PLA 和PBAT 2 者熔点之间对PBAT/PLA共混物进行超临界流体发泡,PLA 可起到物理交联点与非均质成核点的作用,能有效提高共混物低频下的黏度,改善材料的发泡性能。
(2)在PLA 的存在下,通过两步升温法制备出表面光滑不黏结、低收缩率的PBAT/PLA 发泡珠粒。
猜你喜欢
包装工程(2022年1期)2022-01-26 09:03:10
科教导刊·电子版(2021年6期)2021-05-06 05:05:14
工程塑料应用(2020年11期)2020-11-28 01:57:50
中国科技博览(2017年39期)2017-09-07 09:14:31
核技术(2016年4期)2016-08-22 09:05:24
塑料制造(2016年5期)2016-06-15 20:27:39
中国塑料(2016年7期)2016-04-16 05:25:46
中国塑料(2015年2期)2015-10-14 05:34:14
合成材料老化与应用(2015年4期)2015-07-25 10:45:44
中国光学(2015年4期)2015-05-12 21:55:09
