
N掺杂TiO2纳米管阵列的制备及光电化学检测性能的研究*
2022-11-01 13:18杨振宇沈子函徐光青崔接武吴玉程
功能材料 2022年10期
杨振宇,沈子函,徐光青, 2,吕 珺, 2,崔接武, 2,吴玉程, 2
(1. 合肥工业大学 材料科学与工程学院, 合肥 230009;2. 合肥工业大学 先进功能材料与器件安徽省重点实验室, 合肥 230009)
0 引 言
近年来,随着社会的发展,水体污染的问题也越来越严重。目前已知的有机物种类达700多万种,这些有机物随着污水排放进入各种水体环境中,对人们的身体健康产生极大的影响[1]。传统检测方法,如库伦滴定法等,均使用重铬酸钾作为氧化剂,对水体中有机物进行氧化并获得其化学需氧量的值。但传统的重铬酸钾法面临检测效率低,对复杂环境中水体所含有机物不敏感,以及所使用的重铬酸盐对水体存在二次污染等问题。随着电化学氧化[2]及光催化氧化[3-4]的出现,使得有机物检测取得了突破。但电化学氧化法虽然具备快速、灵敏度高及无二次污染等优点,也面临检测效率低,检测极限高等缺点。同样,光催化氧化法具备氧化速率快,环保等优点,但表面易复合的光生电子-空穴对,使得材料自身的光催化活性受到限制,从而影响其有机物检测性能[5]。进一步的研究发现将光催化氧化与电化学氧化结合在一起的光电化学氧化,可以综合二者的优点。施加的外部电场可以抑制光催化材料表面光生电子-空穴对的复合,实现更加有效的载流子分离,提高其光催化活性。其次,光生电子通过外电路形成电信号,能够快速的获得样品的电化学信号,实现对有机物的快速响应。这使得选用的材料不仅需要满足光催化活性的要求,还需要具备一定的导电性。TiO2纳米管阵列是目前应用最广泛的光催化材料,自从1972年Fujishima等发现其在光解水方面的应用以来,因具备较好的光催化活性、优异的化学稳定性、低成本及无毒无害等优点引起人们的关注[6]。TiO2表面的氧化反应过程通常存在两种,一种是空穴(h+)与OH-反应生成羟基自由基(·OH),羟基自由基(·OH)与溶液中的有机物再进行反应,其次是空穴(h+)与溶液中有机物直接氧化反应。在水的氧化和有机物氧化这一对竞争性反应中,如何有效地降低背景光电流,增加有机物响应电流成为人们研究的重点。
TiO2纳米阵列因其禁带宽度大,电子空穴复合几率高等问题而在实际应用方面受到限制。针对这些问题,人们提出了不同的改性方法,如掺杂[7-8],调控晶面[9],半导体复合[10-11]及有机物敏化[12]等。其中,相较于其他方法,掺杂是唯一能够提升TiO2本征光响应能力和改善本征电导特性的改性方法。按掺入元素的不同分为金属掺杂及非金属掺杂,金属掺杂是将金属离子引入TiO2晶格中,取代Ti4+,这种方法因其成本较高且大部分以溶胶凝胶法进行制备,实际应用受到很大的限制[13-14],而非金属掺杂是将非金属原子引入晶格中,取代TiO2中的O原子或者将非金属原子引入晶格间的间隙中,不仅能够拓宽TiO2的光吸收范围,还可以降低电子-空穴复合几率。Praveen等通过将TiO2NTAs的阳极氧化制备及退火过程均在N2气氛中进行,获得了N掺杂的TiO2NTAs,禁带宽度降至2.7 eV[15]。Liu等将TiO2NTs和PVA及尿素一起在N2气氛中退火获得C/N共掺杂的TiO2NTs,禁带宽度同样降低至2.64 eV[16]。光吸收范围的增加及光生载流子复合几率的降低,使得样品产生的空穴(h+)浓度增加。通过非金属掺杂,在TiO2价带与导带之间引入了新的杂质能级,价带位置出现负移,减弱了样品光解水能力,增强了样品表面空穴(h+)的直接氧化作用,这有利于改善TiO2NTAs的光电化学检测有机物性能。
目前非金属掺杂工艺,大部分局限于TiO2纳米粉的掺杂,块状TiO2NTAs样品主要是以离子注入[17]、热处理[18]、气相沉积法[19]、电化学法[20]及液相法[21]进行掺杂。其中离子注入、热处理、气相沉积及电化学法等方法,不仅会增加工艺所需要的成本,而且这些方法处理后会对TiO2NTAs的形貌结构产生较大的影响,这同样不利于样品的光响应性能。
本文采用阳极氧化结合液相法,以二乙烯三胺为N源,通过20~80 ℃不同温度下浸渍一段时间并在N2气氛中退火,成功制备得到N掺杂TiO2NTAs,研究了其对有机物的光电化学检测性能。
1 实 验
1.1 试剂与仪器
制备TiO2NTAs所用原材料为0.25 mm厚的钛箔(99.7%)。丙酮、无水乙醇、乙二醇((CH2OH)2)、氟化铵(NH4F)、二乙烯三胺(C4H13N)和硫酸钠(Na2SO4)均来自阿拉丁试剂(中国上海),化学试剂纯度均为分析纯。溶液配置过程中的水均使用去离子水(18 MΩ/cm)。
荷兰帕纳科公司PANalytical X-Pert PRO MPD型X射线衍射仪(XRD),Cu Kα射线;日本日立SU8020型冷场发射扫描电子显微镜(SEM);美国Thermo公司ESCALAB250Xi型X射线光电子能谱仪(XPS),Al Kα射线;美国Thermo公司Nicolet型傅里叶红外光谱仪(FTIR);法国HORIBA JOBIN YVON公司LabRAM HR Evolution型显微共焦激光拉曼光谱仪(Raman);美国安捷伦公司CARY 5000紫外可见近红外分光光度计(UV-Vis);日本日立公司F-4500型荧光分光光度计(PL)。
1.2 实验过程
1.2.1 TiO2NTAs阵列的制备
将Ti片剪成直径为14 mm的圆片,并将其依次在丙酮、乙醇及去离子水中分别超声清洗30 min,随后将Ti片放在鼓风干燥箱中进行干燥。将2.83 g氟化铵及25 mL去离子水加入475 mL乙二醇中,室温下搅拌至完全溶解后,获得所需要的阳极氧化电解液。阳极氧化过程采用恒压直流电源系统的两电极系统中进行,Ti片为正极,石墨片为负极,氧化电压为60 V,氧化时间为6 h。阳极氧化结束后,在乙二醇中超声2 min以去除表面破碎的TiO2纳米管,获得TiO2NTAs。将清洗后的TiO2充分干燥后放入马弗炉中,空气气氛下,以1 ℃/min速率升温至500 ℃并保温2 h。随炉冷却后即可获得所需要的锐钛矿相TiO2NTAs。
1.2.2 N掺杂TiO2NTAs的制备
将上述锐钛矿相TiO2NTAs放入30 mL二乙烯三胺溶液中,超声1 min,在20,40,60和80 ℃不同温度下分别静置4 h,随后取出并用去离子水冲洗样品表面,放入干燥箱中干燥。将干燥后的样品放入管式炉中,在N2气氛下,以1℃/min升温至400 ℃并保温2 h后随炉冷却至室温,获得N掺杂TiO2NTAs,按静置温度将其依次命名为TiO2(N20) NTAs、TiO2(N40) NTAs、TiO2(N60) NTAs和TiO2(N80) NTAs。作为对比,40 ℃下将样品浸渍在去离子水中4 h,并在N2中进行退火,命名为TiO2(N2) NTAs。
1.2.3 光电化学性能测试
采用CHI660D电化学工作站对样品的光电化学性能进行测量。测试所使用的电解液为含0.5 mol/L Na2SO4的水溶液,所用紫外光源为波长365 nm的LED紫外光源(光斑直径10 mm,光强为48 mW/cm2),以带420 nm截断滤波片的500 W Xe灯作为可见光源(光强为100 mW/cm2)。在0.2 V下,每隔100 s开关光一次,获得的电流-时间曲线即为样品的光响应电流曲线。同样在施加0.2 V偏压的情况下,测量了样品的EIS电化学交流阻抗谱(频率范围为0.1 Hz至 100 000 Hz,调制幅度为10 mV)。采用电化学工作站测试样品的Mott-Schottky曲线,扫描范围为-1~1 V,交流频率为1 kHz。
以葡萄糖为目标有机物,采用365 nm固定波长的LED紫外灯作为光源,恒电位模式下进行样品的有机物响应电流测试。在样品光电流稳定后,每隔100 s向50 mL缓冲溶液中滴加50 μL的葡萄糖溶液(葡萄糖溶液浓度为0.1 mol/L),获得响应光电流与有机物浓度的对应关系,测试其灵敏度、检测极限以及光电化学传感性能。
高中物理知识有其自身的规律,在进行探究式教学的时候,教师需要根据教材内容和学生的认知能力进行合理的引导,让学生能够在探究中发现物理知识,感受物理知识的生成过程,从而提高学生的自主探究能力.教师要根据物理知识的内在规律,引导学生进行假设和猜想,并运用实验的方法进行猜想的验证,教师对学生进行正确的引导,可以让学生逐步的掌握物理知识探究方法,提高学生的物理自主探究能力.
2 结果与讨论
2.1 样品的表征
图1为TiO2NTAs和不同条件得到的TiO2(N) NTAs样品的X射线衍射(XRD)图谱以及扫描电子显微(SEM)图片。由图1(a)可知,2θ为25.3°、37.8°和48.0°的衍射峰分别对应锐钛矿相TiO2的(101)、(004)和(200)晶面,而在40.2°、53.0°和63.0°的衍射峰则来源于Ti基底[20]。N掺杂后TiO2对应的衍射峰并未出现明显变化,说明N掺杂并未使得TiO2NTAs表面出现新相或者引起TiO2的相结构变化。N掺杂前后TiO2NTAs的SEM形貌分别示于图1(b)和 (c)中。可知TiO2NTAs的管径大约为110 nm,壁厚约20 nm,N掺杂对TiO2NTAs的形貌和尺寸无明显影响。

图1 样品的XRD图谱(a)以及SEM图片:(b) TiO2 NTAs,(c) TiO2(N40) NTAsFig.1 XRD patterns (a), SEM images of (b) TiO2 NTAs and (c) TiO2(N40) NTAs
图2为TiO2NTAs、TiO2(N2) NTAs和TiO2(N) NTAs样品的X射线光电子能谱(XPS),其中图2(a)为总谱图,可以看出不同样品中主要元素均为Ti、O、N及C,其中C元素是为了进行荷电校正而引入的吸附碳。图2(b)为Ti 2p的高分辨率图谱,其中位于458.7及464.4 eV处的特征峰分别为Ti 2p3/2及Ti 2p1/2电子的结合能峰,归属于Ti4+[22-23]。N掺杂后的Ti 2p高分辨率图谱中的Ti 2p3/2及Ti 2p1/2电子的结合能峰与原样相比几乎都没有移动,原因可能是N的掺杂量太少或者N进入TiO2晶格中,并没有取代O而可能以间隙的形式存在于TiO2晶格间,对Ti4+周围的电子云影响较小,使得Ti 2p的结合能峰的位置没有发生明显的变化。图2(c)为O 1s电子的高分辨率图谱,529.9 eV处的特征峰对应与TiO2中的O,而531.4 eV处的特征峰对应样品表面吸附的羟基(-OH)[24]。N掺杂前后,样品中O的结合能峰的位置也没有发生变化。图2(d)为N 1s高分辨率图谱,如图2(d)所示,TiO2NTAs和TiO2(N2) NTAs样品的N高分辨谱中只存在400.1 eV处的特征峰,来源于样品在表面中吸附的N2,而TiO2(N) NTAs样品在拟合后均出现了399.6 eV处的特征峰,这是间隙N原子在样品表面存在的标志,说明掺杂的N原子是以间隙的形式存在于TiO2的晶格之中[25]。表1为TiO2NTAs、TiO2(N2) NTAs以及不同条件下TiO2(N) NTAs样品的N含量,TiO2(N) NTAs表面均存在间隙N原子,且TiO2(N40) NTAs样品的间隙N原子含量最高,为3.14 %(原子分数)。

图2 TiO2 NTAs, TiO2(N2) NTAs和TiO2(N) NTAs的XPS图谱:(a)总谱图;(b) Ti 2p;(c)O 1s;(d)N 1sFig.2 XPS patterns of TiO2 NTAs, TiO2(N2) NTAs and TiO2(N) NTAs: (a) survey patterns; (b) Ti 2p; (c) O 1s; (d) N 1s

表1 TiO2 NTAs, TiO2(N2) NTAs和TiO2(N) NTAs的N含量Table 1 N atomic contents of TiO2 NTAs, TiO2(N2) NTAs and TiO2(N) NTAs
图3为TiO2NTAs和TiO2(N40) NTAs样品的拉曼光谱(a)及傅里叶红外光谱(b)。图3(a)中,TiO2NTAs的拉曼光谱中在139,400,520及642 cm-1处的特征峰分别对应TiO2的Eg、B1g、B1g或A1g和Eg模式。与TiO2NTAs相比,TiO2(N40) NTAs样品的特征峰分别移至143,403,522及644 cm-1。这是由于N原子进入TiO2晶格间,导致了晶格间应力的产生,使得拉曼光谱出现轻微蓝移的现象。由于N—H基团以及Ti—N基团拉曼活性低,在拉曼光谱中并未观察到明显的N-H键及Ti-N键的振动。样品在1 300~1 600 cm-1之间没有出现新的峰,则说明制备过程中使用的胺等有机物,并未在退火过程中碳化引入杂质元素C。图3(b)为TiO2NTAs和TiO2(N40) NTAs样品的傅里叶红外光谱,图中804 cm-1处的峰对应Ti-O键的拉伸振动[26],与TiO2NTAs相比,TiO2(N40) NTAs在此处的峰较为平坦,说明N原子的掺入破坏了Ti-O键的振动。1 034 cm-1处的峰是由于N元素进入TiO2晶格间隙产生的Ti-N键的振动[27],但是740 cm-1处却未出现Ti-N的峰,这证明N原子没有取代O原子,与Ti原子成键形成TiN[28]。因此,N原子是以间隙N的形式存在于TiO2NTAs的晶格之中,这与图2(d)中分析得到的结果相对应。在1 588 cm-1及2 859~2 921 cm-1处的峰则分别对应于由于浸渍过程中二乙烯三胺分解产生的N—H键的扭曲振动及拉伸振动[27,29]。
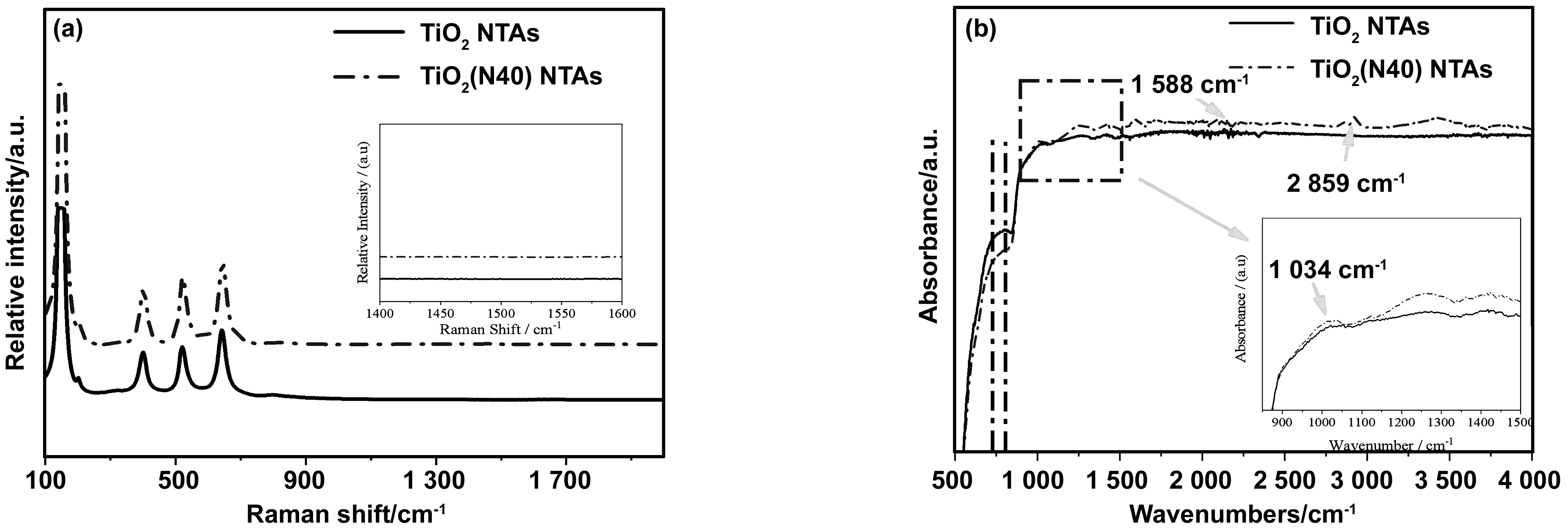
图3 TiO2 NTAs和TiO2(N40) NTAs的拉曼光谱(a)以及傅里叶红外光谱(b)Fig.3 Raman spectra (a) and FT-IR spectra (b) of TiO2 NTAs and TiO2(N40) NTAs
2.2 光电化学性能
图4为TiO2NTAs、TiO2(N2) NTAs和TiO2(N) NTAs样品的电流-时间曲线图。图4(a)为样品的紫外光响应电流-时间曲线,TiO2NTAs的紫外光电流约为180.4 μA,TiO2(N2) NTAs的紫外光响应电流与原样相近,而N掺杂后的TiO2(N) NTAs样品的紫外光响应电流均得到明显提升。20,40,60及80 ℃温度下浸渍的TiO2(N) NTAs样品的紫外光光电流依次为229.9,256.8,244.9及208.1 μA。最优化TiO2(N40) NTAs样品的紫外光响应电流增至256.8 μA,约为原样的1.42倍。图4(b)为样品的可见光响应电流-时间曲线。未掺杂TiO2NTAs的可见光响应电流仅为0.9 μA,TiO2(N2) NTAs的光电流略有上升,约为1 μA。N掺杂后可见光响应电流均有提升,20、60及80 ℃温度下的TiO2(N) NTAs样品的可见光响应电流依次为1.7,1.5及1.3 μA。与紫外光响应电流的结果相对应的是,在浸渍温度为40 ℃的情况下,可见光响应电流达到最大值2.8 μA,为TiO2NTAs可见光电流的3倍。由上述可知,最优化样品为TiO2(N40) NTAs,这与上述表1中TiO2(N40) NTAs的间隙N原子量最高相对应,说明随着N掺入量的提高,光电化学性能得到了明显的提升。

图4 TiO2 NTAs, TiO2(N2) NTAs以及TiO2(N) NTAs的紫外(a)以及可见(b)光电流Fig.4 Ultraviolet (a) and visible (b) photocurrents of TiO2 NTAs, TiO2(N2) NTAs and TiO2(N) NTAs
图5为基于TiO2NTAs和TiO2(N40) NTAs样品的光电化学传感器对有机物的响应电流。图5(a)为TiO2NTAs和TiO2(N40) NTAs样品的光电流-时间曲线。TiO2NTAs和TiO2(N40) NTAs样品在缓冲电解液中的基础光电流分别为174.6和264.2 μA。随着有机物浓度的增加,光电流均呈台阶式上升。由图5(a)可知,每滴加50 μL的0.1 mol/L有机物溶液,TiO2NTAs样品的光电流响应为6 μA,而TiO2(N40) NTAs样品的光电流响应增加至12 μA。图5(b)为TiO2NTAs和TiO2(N40) NTAs样品的连续响应曲线,为了方便进行比较,对TiO2NTAs及TiO2(N40) NTAs样品的有机物连续响应曲线作归一化处理,即将在缓冲电解液中的基础光电流设为零点。由图可知,随着有机物浓度的增加,光电流响应先逐渐增加直至不在明显变化。与TiO2NTAs原样相比,TiO2(N40) NTAs样品对有机物的响应光电流曲线增加的更加明显且更加稳定。通过放大第一阶段的电流-时间曲线,获得图5(c)所示的检测噪音,TiO2NTAs样品的检测噪音约0.736 μA,而TiO2(N40) NTAs样品的检测噪音约为0.609 μA。图5(d)为电流增量与有机物浓度之间的关系图,拟合后得到的直线斜率即为所需要的检测灵敏度。通过公式dl=3σ/m(其中σ为图5(c)中的检测噪音,m为图5(d)中拟合得到直线斜率)计算得到检测极限。TiO2NTAs的检测灵敏度为0.061 μA/(μmol/L), 检测极限为36.20 μmol/L,而TiO2(N40) NTAs的检测灵敏度增加至0.134 μA/(μmol/L),检测极限降至13.63 μmol/L。更高的灵敏度和更低的检测极限意味着更高的传感器检测性能。

图5 基于TiO2 NTAs和TiO2(N40) NTAs样品的光电化学传感器对有机物响应性能的测试:(a,b)连续滴加有机物的电流-时间曲线;(c)检测噪音;(d)电流增量与有机物浓度之间的关系图Fig.5 (a), (b) Amperometric response of obtained TiO2 NTAs and TiO2(N40) NTAs PEC sensor to successive additions of glucose in 0.5 mol/L buffer solution, (c) detect noise, and (d) plots of current increments vs. organic concentration range
2.3 光电化学机理分析
通过上述分析可知,N掺杂TiO2NTAs具有较高的光电化学检测性能,其机理将从能带结构和电化学性能方面进行分析。图6所示为TiO2NTAs和TiO2(N40) NTAs样品的紫外-可见光吸收光谱、XPS价带谱以及基于两者得到的能带结构示意图。图6(a)为TiO2NTAs和TiO2(N40) NTAs样品的紫外-可见光光吸收谱。由图可以看出,N掺杂后TiO2NTAs在吸收边末端出现吸收肩,说明N的掺入,拓展了TiO2NTAs的光响应范围,降低了禁带宽度。通过Kubelka-Munk公式换算,TiO2(N40) NTAs的禁带宽度约为3.08 eV,低于TiO2NTAs (3.22 eV)。图6(c)为TiO2NTAs和TiO2(N40) NTAs样品的价带谱,TiO2NTAs样品的价带位置在2.56 eV,而TiO2(N40) NTAs样品则移至2.43 eV。由上述结果可以得到如图6(d)所示的能带结构示意图。TiO2(N) NTAs中存在表面掺杂部分和内部未掺杂部分构成的同型异质结构。TiO2NTAs导带位置与TiO2(N) NTAs的导带位置相近,而价带位置则由TiO2NTAs的2.56 eV移至2.43 eV,意味着空穴(h+)可以由TiO2NTAs的价带迁移至表面TiO2(N) NTAs的价带处。这不仅促进了光生载流子的分离,同时空穴迁移至样品表面,基于空穴(h+)对有机物的直接氧化以及羟基自由基(·OH)对有机物的氧化作用,TiO2(N40) NTAs样品对有机物的氧化性能明显提升,光电化学检测性能得到明显的改善。

图6 TiO2 NTAs和TiO2(N40) NTAs样品的紫外可见光谱(a)及转换的Kubelka-Munk函数和光子-能量曲线(b); (c) TiO2 NTAs及TiO2(N40) NTAs的价带谱; (d)能带结构示意图Fig.6 (a) UV-Vis spectra of TiO2 NTAs and TiO2(N40) NTAs and (b) photon-energy curves obtained by Kubelka-Munk function, (c) valence band spectrum and (d) schematic diagram of band structure
通过在缓冲电解液中加入相应自由基的捕获剂来调控样品表面的电化学反应过程,依此探究N掺杂对样品光电化学检测性能的影响。图7所示为TiO2NTAs及TiO2(N40) NTAs样品在缓冲电解液、含50 μmol/L异丙醇和含50 μmol/L草酸铵缓冲电解液中的紫外光响应电流。异丙醇能够捕获样品光解水产生的羟基自由基(·OH),而草酸铵能够捕获样品表面的空穴(h+)。图7(a)中,TiO2NTAs在缓冲电解液、含50 μmol/L异丙醇和含50 μmol/L草酸铵缓冲电解液中的紫外光响应电流分别为180.3,401.1及383.2 μA,说明样品表面间接氧化对空穴(h+)的消耗接近于空穴(h+)总消耗量的全部,这不利于样品的光电化学检测性能。而TiO2(N40) NTAs样品在缓冲电解液、异丙醇和草酸铵缓冲电解液中的紫外光响应电流分别为256.8,483.3以及550.0 μA。TiO2(N40) NTAs样品间接氧化消耗空穴(h+)的量占据空穴(h+)总消耗量的87.9 %(这是由483.3 μA除以550.0 μA得到的),空穴(h+)的直接氧化占据12.1 %,说明N掺杂改变了TiO2NTAs表面的电化学反应过程。空穴(h+)直接氧化所占空穴(h+)总消耗量比例的提升,有利于样品光电化学检测有机物性能的改善。

图7 TiO2 NTAs(a)和TiO2(N40) NTAs(b)分别在缓冲电解液、草酸铵(AO)及异丙醇(IPA)溶液中的紫外光响应电流Fig.7 Photocurrents of TiO2 NTAs(a) and TiO2(N40) NTAs(b) in buffer, IPA and AO solutions
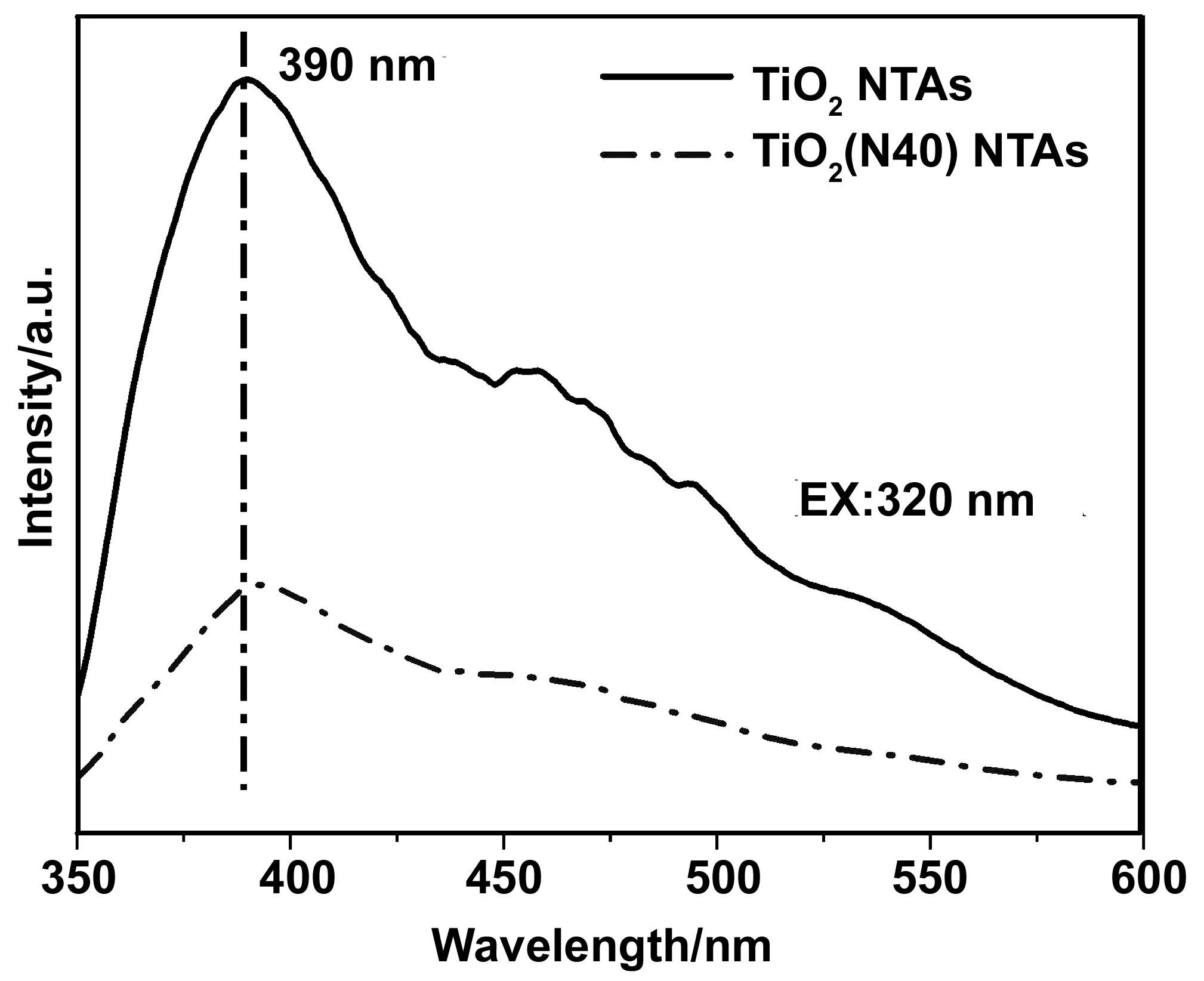
图8 TiO2 NTAs和TiO2(N40) NTAs样品的光致发光图谱Fig.8 PL spectra of TiO2 NTAs and TiO2(N40) NTAs
图9为TiO2NTAs和TiO2(N40) NTAs的电化学阻抗(EIS)图谱以及Mott-Schottky曲线。图9(a)和(b)分别对应TiO2NTAs和TiO2(N40) NTAs样品在光照及暗态条件下的电化学阻抗(EIS)图谱。圆弧的半径对应于样品的电荷转移电阻,半径越小,说明电荷传递过程中遇到的阻抗越小。可以看出,无论有无光照的情况,TiO2(N40) NTAs样品的EIS圆弧半径均小于TiO2NTAs的半径。受光激发的情况下,光生载流子在TiO2(N40) NTAs样品中迁移速率比在TiO2NTAs中更快。图9(c)为光照条件下的伯德相图,TiO2(N40) NTAs特征峰值为0.63 Hz,与TiO2NTAs在52.48 Hz处的特征峰值相比下降的十分明显,表明电子-空穴对在TiO2(N40) NTAs中复合速率降低,载流子的寿命更长。通过下列公式可以计算得到载流子的寿命[30]:
其中fmax为图9(c)伯德相图中得到的特征峰值。由计算得出,TiO2NTAs中载流子的寿命约为30 ms,而TiO2(N40) NTAs的载流子寿命约在252.63 ms,说明N的掺入使得电子-空穴对复合速率降低从而提高光生载流子寿命。图9(d)为TiO2NTAs和TiO2(N40) NTAs样品的Mott-Schottky曲线,TiO2NTAs作为典型的N型半导体,Mott-Schottky曲线的斜率为正值。半导体的载流子密度与Mott-Schottky曲线中的斜率成反比,可知,TiO2(N40) NTAs的斜率明显小于TiO2NTAs,说明TiO2(N40) NTAs具有更高的载流子密度[31]。
由上述分析结果可知,N掺杂降低了样品中的光生电子-空穴对的复合几率,延长了载流子的寿命。较高的载流子分离速率以及载流子在样品中的快速迁移,使得制备得到的TiO2(N) NTAs样品的光响应电流得到提高,光响应性能得到改善。空穴(h+)由TiO2(N) NTAs的体内未掺杂部分迁移至表面掺N部分,改变了样品表面的电化学反应过程,使得TiO2(N40) NTAs样品的光电化学检测有机物性能得到明显的提升。
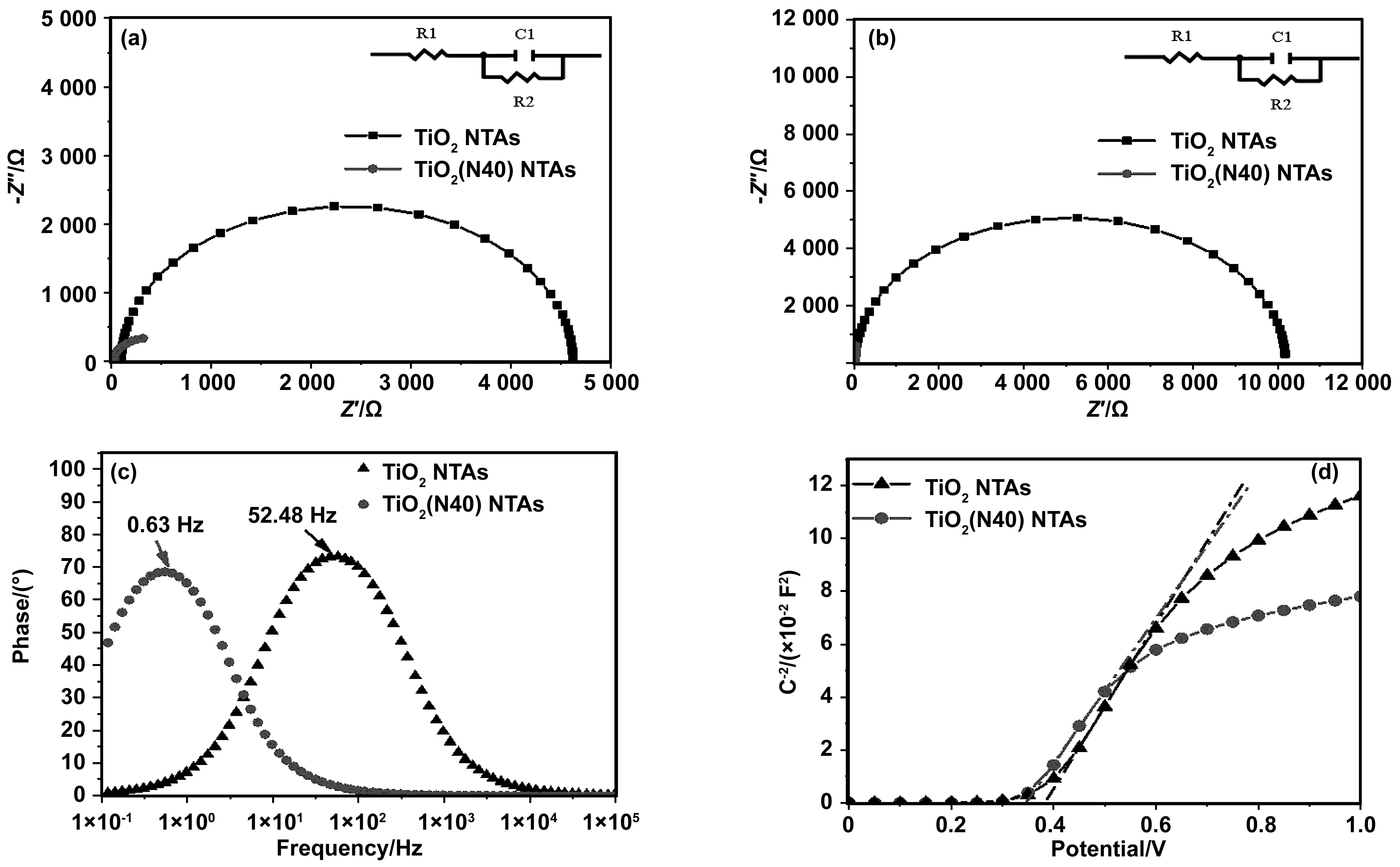
图9 TiO2 NTAs和TiO2(N40) NTAs样品的EIS奈奎斯特图:光照(a);暗态(b);(c)伯德相图;(d) TiO2 NTAs和TiO2(N40) NTAs样品的Mott-Schottky曲线Fig.9 Electrochemical impedance spectroscopy Nyquist plots of TiO2 NTAs and TiO2(N40) NTAs: (a) under illuminate; (b) in dark; (c) Bode phase plots; (d) Mott-Schottky curves of TiO2 NTAs and TiO2(N40) NTAs
3 结 论
通过阳极氧化结合液相处理技术成功制备了N掺杂TiO2NTAs,并对其光电化学性能和有机物光电化学检测性能进行研究。研究结果如下:
(1)通过阳极氧化结合液相处理技术所制备的TiO2(N) NTAs中,N原子以间隙N的形式存在于TiO2的晶格间隙中。其中TiO2(N40) NTAs间隙N最高达到3.14 %(原子分数)。
(2)非金属N掺杂可提升TiO2NTAs的紫外和可见光电化学性能,最优化情况下的TiO2(N40) NTAs的紫外光响应电流由未掺杂TiO2NTAs的180.4 μA提升至256.8 μA。N掺杂也可以提升TiO2NTAs对有机物的光电化学检测性能,TiO2(N40) NTAs的检测灵敏度由0.061 μA/(μmol/L)提升至0.134 μA/(μmol/L),最低检测限由36.20 μmol/L降低至13.31 μmol/L。
(3)机理分析表明,表面非均相N的掺杂,降低了样品的禁带宽度,并使得TiO2(N) NTAs内部未掺杂部分与表面掺杂部分构成同型异质结构。光生载流子的有效分离以及快速的迁移,提高了TiO2(N) NTAs样品的光电化学性能。
猜你喜欢
宇航计测技术(2022年4期)2022-09-07
物理学报(2022年6期)2022-03-30
教学考试(高考物理)(2020年2期)2020-11-13
物理教学探讨(2020年8期)2020-09-22
物理学报(2019年16期)2019-08-29
物理学报(2019年4期)2019-03-16
科技风(2018年9期)2018-05-14
优雅(2018年3期)2018-03-08
江苏农业科学(2017年13期)2017-09-28
中学物理·高中(2017年5期)2017-06-19
