
COVID-19 and hepatorenal syndrome
2022-10-24 09:10HenryWuVarinderAthwalPhilipKalraRajkumarChinnadurai
World Journal of Gastroenterology 2022年39期
Henry H L Wu, Varinder S Athwal,Philip A Kalra,Rajkumar Chinnadurai
Abstract Coronavirus disease 2019 (COVID-19) is a highly infectious disease which emerged into a global pandemic. Although it primarily causes respiratory symptoms for affected patients, COVID-19 was shown to have multi-organ manifestations. Elevated liver enzymes appear to be commonly observed during the course of COVID-19, and there have been numerous reports of liver injury secondary to COVID-19 infection. It has been established that patients with pre-existing chronic liver disease (CLD) are more likely to have poorer outcomes following COVID-19 infection compared to those without CLD. Co-morbidities such as diabetes, hypertension, obesity, cardiovascular and chronic kidney disease frequently co-exist in individuals living with CLD, and a substantial population may also live with some degree of frailty. The mechanisms of how COVID-19 induces liver injury have been postulated. Hepatorenal syndrome (HRS) is the occurrence of kidney dysfunction in patients with severe CLD/fulminant liver failure in the absence of another identifiable cause, and is usually a marker of severe decompensated liver disease. Select reports of HRS following acute COVID-19 infection have been presented, although the risk factors and pathophysiological mechanisms leading to HRS in COVID-19 infection or following COVID-19 treatment remain largely unestablished due to the relative lack and novelty of published data. Evidence discussing the management of HRS in highdependency care and intensive care contexts is only emerging. In this article, we provide an overview on the speculative pathophysiological me-chanisms of COVID-19 induced HRS and propose strategies for clinical diagnosis and management to optimize outcomes in this scenario.
Key Words: COVID-19; Hepatorenal syndrome; Pathophysiology; Clinical assessment;Management; Prognosis
lNTRODUCTlON
The impact of coronavirus disease 2019 (COVID-19) has been tremendous since the initial case was reported in December 2019, and COVID-19 subsequently spiraled into a global pandemic which is affecting populations and societies significantly up to this day[1-3]. The severity of COVID-19 could be wide ranging from mild to severe disease[4]. This depends on various intrinsic and environmental factors for each individual[4,5]. The manifestations of COVID-19 are thought to be primarily respiratory,with severe COVID-19 infection leading to acute respiratory distress syndrome potentially progressing towards life-threatening septic shock and multi-organ failure[6,7]. There is emerging evidence on the multi-systemic effects of COVID-19 outside of the respiratory system. It is suggested that COVID-19 has direct associations with acute disease processes across the neurological, cardiovascular, renal and gastroenterological systems amongst other organ systems, but the pathophysiology of how COVID-19 affects these organs has not been fully established in most instances[8-12].
Liver injury secondary to COVID-19 has been investigated, with its incidence ranging between 15%and 53%[13]. In most patients, the effect of COVID-19 on the liver is a transient reaction with elevation of transaminases which resolves and most patients achieve recovery back to their normal baseline[14].However, individuals with underlying cirrhosis and chronic liver disease (CLD) were found to have significantly greater 30-day mortality and lengthier hospitalization, and poorer prognosis following acute recovery from COVID-19 induced liver injury[15]. Histological damage to hepatocytes and bile duct cells was found in patients testing positive for COVID-19[16].
Hepatorenal syndrome (HRS) is a state of kidney function deterioration (usually profound oliguria and sodium retention) in patients with advanced cirrhosis or acute liver failure[17,18]. The decline in kidney function could be rapid (Type I) or gradual (Type II), dependent on the etiology of HRS[18]. In cirrhosis, Type 1 and Type 2 HRS has been replaced with newer terminology and HRS is now defined as either acute (HRS-AKI), sub-acute (HRS-AKD) or chronic (HRS-CKD)[19]. HRS is a diagnosis of exclusion in which other causes of kidney dysfunction are not identified, and where the kidneys were not found to be structurally damaged[17,18]. The dominant theory to explain for the pathophysiology of HRS is significant constriction of blood vessels which perfuse the kidneys, most likely mediated by splanchnic vasodilation (leading to central hypovolemia) and hepatorenal reflex mechanisms as a result of portal hypertension[17,18]. HRS is most likely seen in the context of advanced stage cirrhosis. The most common etiologies of cirrhosis include alcohol, non-alcoholic steatohepatitis and chronic viral hepatitis[20]. Multiple triggers of HRS have been recognized and spontaneous bacterial peritonitis (SBP)in patients with ascites from decompensated cirrhosis is a leading cause[21]. HRS is a marker of poor prognosis in hepatology, and the risk of death is very high unless prompt liver transplantation or acute dialysis can be provided[22].
There were select case reports of liver injury following COVID-19 infection where acute kidney dysfunction was found, suggesting the potential manifestation of COVID-19 induced HRS[23,24].Evidence of the pathophysiology, optimal strategies for clinical assessment and management of COVID-19 induced HRS is seldom discussed at present due to a relative lack of cases. In this review, we will explore the speculative pathophysiological mechanisms of HRS following COVID-19 infection based on early evidence, and propose potential clinical assessment and management strategies to optimize HRS outcomes in this scenario.
POTENTlAL PATHOPHYSlOLOGlCAL MECHANlSMS OF COVlD-19 lNDUCED HRS
Current perspectives on the potential pathophysiological mechanisms of COVID-19 induced HRS are that of a multifactorial process (Figure 1). COVID-19 induced liver injury can be the result of direct viral cytopathic hepatocyte injury, systemic inflammatory cytokine storms causing hepatocyte cell death,endothelitis and dysfunction of the liver vasculature leading to widespread cell damage and ischemia,and drug-associated exacerbations in COVID-19 induced liver injury[25,26]. Subsequently, these multiple pathways of liver injury may result in circulatory dysfunction, progressing to HRS with vasoconstriction and hypoperfusion of the kidneys amongst other mechanisms[17-19].
Direct COVID-19 infection of hepatocytes
Direct viral cytopathic injury to the liver, similar to how various organs are affected by COVID-19 infection, should be considered as the major pathophysiological mechanism. Ultrastructural histological examination identified severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) particles in the cytoplasm of hepatocytes[27]. Features of cellular viral invasion such as conspicuous mitochondrial swelling, decreased glycogen granules, and endoplasmic reticulum dilatation were observed in SARSCoV-2-infected hepatocytes[27]. The presence of binuclear hepatocytes, central lobular necrosis and hepatocyte apoptosis were observed features of liver damage following COVID-19 infection[27].Induction of hepatocyte apoptosis from COVID-19 infection could be the result of p7a overexpression[26,28]. p7a is a protein which can be expressed in cells infected by SARS-CoV-2, and induces apoptosis in cell lines derived from organs including the lungs, liver and kidneysviaa caspase-dependent pathway[29]. This mechanism confirms the pathway of how SARS-CoV-2 can directly attack liver tissues and cause damage.
The extent of viral tropism is typically dependent on the availability of viral receptors at the surface of host cells in specific tissues[30]. The spike (S) protein of SARS-CoV-2 mediates cellular entry of SARSCoV-2. S protein is cleaved by transmembrane serine protease 2/transmembrane serine protease 4 and interacts with the angiotensin converting enzyme 2 (ACE2) protein in host cells specifically[30]. In normal circumstances, ACE2 would only be expressed in bile duct epithelial cells, central hepatic vein and portal vein endothelial cells within the hepatobiliary system, being almost absent in hepatocytes[31]. The level of ACE2 expression in bile duct epithelial cells is comparable to that of alveolar epithelial cells in the lungs (commonly recognized to have the greatest expression of ACE2 in the body)[31]. The compensatory differentiation and proliferation of liver parenchymal cells derived from bile duct cells during diseased states may explain the underlying pathophysiological mechanisms of how SARS-CoV-2 induces liver injury[32]. The degree of ACE2 expression in hepatocytes is regulated by multiple factors.Histological studies in mice and humans identified greater ACE2 expression in subjects with cirrhosis[33]. The degree of hypoxia is also shown to correlate with ACE2 expression in hepatocytes, and notably the affinity of S protein towards the ACE2 receptor is increased with hypoxia due to trypsin activation,trypsin being a protein commonly expressed in liver epithelial cells[34,35]. These findings may explain why the effects of COVID-19 infection would tend to be more severe in patients living with underlying CLD and other hypoxic conditions.
Systemic inflammatory cytokine storms in COVID-19 induced liver injury
Another purported pathway of COVID-19 induced liver injury is an excessive immune response triggered by the virus causing a systemic inflammatory cytokine storm[36]. During a systemic inflammatory cytokine storm stimulated by COVID-19 induced liver injury, complement and interleukin-23(IL-23) are released into the bloodstream, activating kupffer cells and inducing their production of tumor necrosis factor α (TNF-α)[37,38]. As an inflammatory cytokine, TNF-α aggravates the responses of inflammation by upregulating the expression of endothelial cell adhesion molecules and inducing hepatocytes to secrete chemokines[39]. Under the induction of chemokines, CD4+T cells and neutrophils are rapidly recruited to the liver, in which CD4+T cells assist mucosal molecules to promote neutrophil entry into the liver parenchyma[40,41]. Neutrophils directly damage hepatocytes by releasing oxidants and proteases, resulting in cell necrosis[41]. In patients who are severely affected or critically ill following COVID-19 infection, much higher plasma levels of inflammatory cytokines and lower lymphocyte counts were observed[42]. Previous studies note that an increase in IL-6 and IL-10 and a decrease in CD4+T cells were independent risk factors related to severe liver damage, and lymphopenia and C-reactive protein levels were found to be independently associated with the degree of liver injury[43].
The inflammatory storm response from COVID-19 infection is usually mild in the early stages of COVID-19 infection, but patients may clinically deteriorate rapidly if appropriate management of COVID-19 is not administered in a timely manner, with the inflammatory storm response occurring more strongly during the post-viral inflammatory phase[6]. For patients living with decompensated CLD complications, notably ascites, their threshold to develop systemic inflammatory storm responses following COVID-19 infection would even be lower than those without CLD due to underlying proinflammatory risks with SBP[44,45].
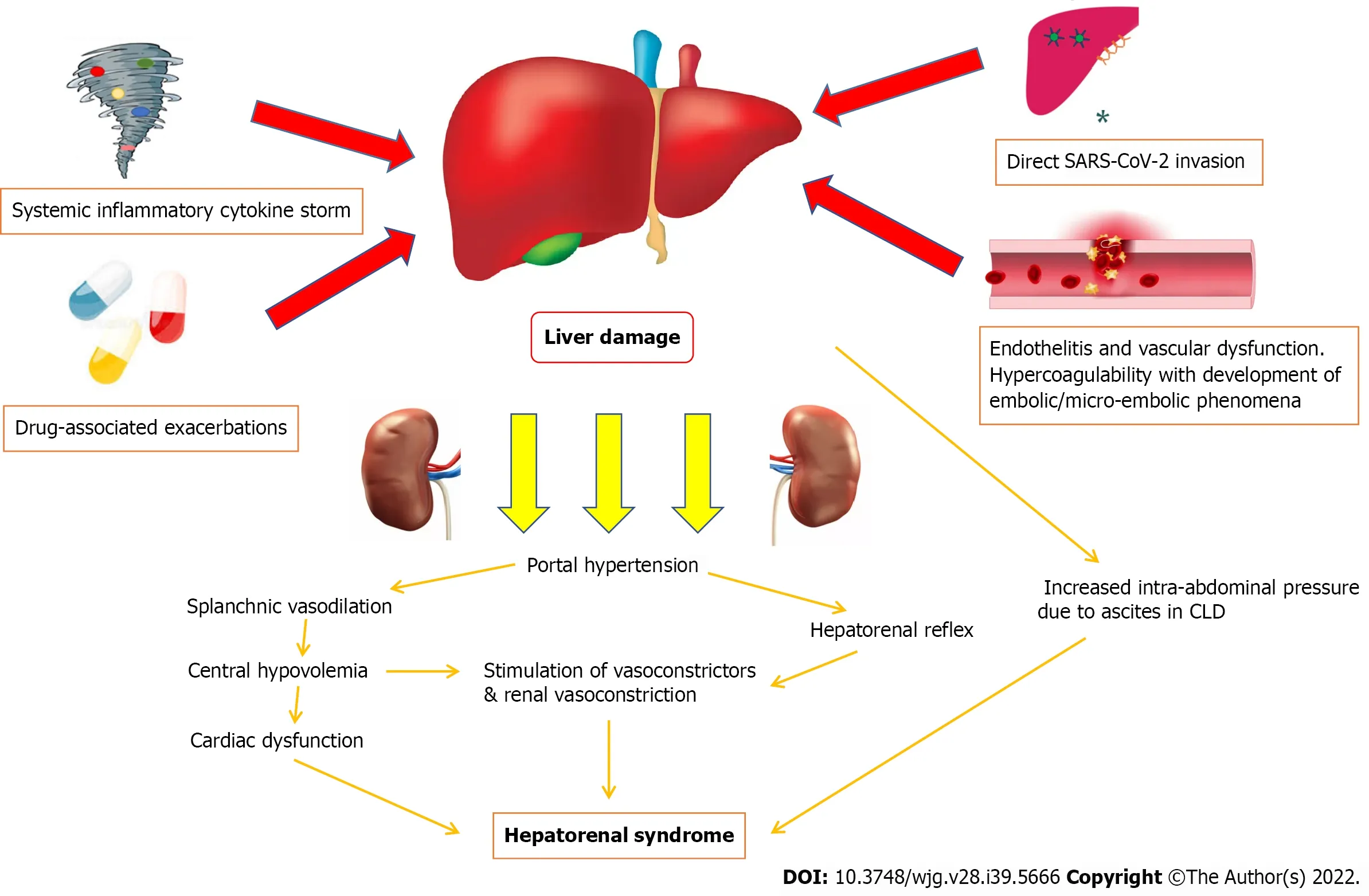
Figure 1 Potential pathophysiological mechanisms of COVlD-19 induced hepatorenal syndrome. SARS-CoV-2: Severe acute respiratory syndrome coronavirus 2.
Endothelitis and vascular dysfunction in COVID-19 induced liver injury
The impact of COVID-19 towards thrombo-inflammation in endothelial tissues is significant. The ACE2 protein, which is present in many organs across the body, facilitates SARS-CoV-2 entry into endothelial cellsviaendocytosis with its binding to the ACE2 protein[46]. Viral infection and immune-mediated inflammatory responses occur within endothelial cells, leading to vascular dysfunction, especially in capillaries[47-49]. Subsequently, vascular dysfunction progresses to a hypercoagulable state and the development of embolic/micro-embolic phenomena, tissue edema, and organ ischemia[47-49]. In the liver, ischemia reperfusion injury which occurs typically after rapid recovery of blood circulation following events of vascular dysfunction leading to ischemia, has been touted as an underlying pathophysiological mechanism of COVID-19 induced liver injury[48,49]. Reperfusion following ischemia activates neutrophils, kupffer cells, and platelets within the cellular surroundings, leading to a series of destructive cellular reactions such as reactive oxygen species and calcium overload, which manifests towards widespread inflammatory response and cell damage[48,49]. Eventually, increased anaerobic glycolysis leads to reduced adenosine triphosphate production, which ultimately results in hepatocyte cell death from inhibition of hepatocyte signal transduction[42,48,49].
Drug-associated exacerbations in COVID-19 induced liver injury
In addition to the direct viral and systemic inflammatory mechanisms of liver injury following COVID-19 infection, the impact of various drugs received during acute hospitalization in patients with COVID-19 associated liver injury has been discussed. There has been debate whether these drugs (drugs trialed/used for COVID-19 treatment-antivirals such as Lopinavir/Ritonavir, Remdesivir, Favipiravir,Arbidol, Oseltamivir and others; antibiotics such as Doxycycline and Azithromycin; chloroquines;steroids; non-steroidal anti-inflammatory drugs) play a greater pathophysiological role in causing liver injury compared to COVID-19 infection itself[50].
Several studies have evaluated antivirals targeting COVID-19 infection in the midst of liver injury.For example, Lopinavir is a protease inhibitor conventionally used to treat human immunodeficiency virus infection in combination with a low dose of Ritonavir, another protease inhibitor, which enhances its biological half-life[51]. If high doses of Ritonavir (> 1 g daily) are taken, severe hepatotoxicity may ensue[52]. Recent studies show that Lopinavir/Ritonavir, when prescribed with or without ribavirin,interferon beta, and/or corticosteroids, was independently associated with increased levels of serum alanine transaminase (ALT) and aspartate transaminase (AST) in patients with positive COVID-19 status[53]. It was demonstrated from a retrospective observational study by Jiang and colleagues that Lopinavir/Ritonavir use in COVID-19 patients is associated with liver injury and abnormal liver function, particularly for patients in a non-critical state[54]. Ultimately, considering the fact that there is yet to be a completely effective antiviral therapy for COVID-19 and that antiviral drugs may cause abnormal liver function, there should be careful consideration of whether to prescribe antivirals, in particular for patients with CLD and/or metabolic diseases[52].
Antibiotics, particularly those of tetracycline-class and Azithromycin, have been shown to exacerbate liver damage in the context of COVID-19 induced liver injury[55]. For example, Doxycycline chelates zinc, which is required by the matrix metalloproteinases involved in COVID-19 infection, and inhibits SARS-CoV-2 RNA polymerase activity and direct viral entry[56,57]. Whilst generally safe to use with its anti-inflammatory effects, Doxycycline use may contribute towards hepatotoxicity and has been linked to occasional bile duct injuries[55]. There should be caution when prescribing Doxycycline alongside other potential hepatotoxic drugs, given reports of fulminant liver failure and hepatocellular necrosis occurring following the prescription of Doxycycline in these scenarios[55].
At a molecular level, there has been focused discussion on cytochrome P450 (CYP450) in the context of drug-associated exacerbations in COVID-19 induced liver injury. CYP450 is a superfamily of monooxygenase enzymes that mediate drug interactions during various pathological conditions[58]. It is presumed the metabolic activity of CYP450 would be altered by the effects of acute COVID-19 infection[57]. Liver injury in the context of COVID-19 infection complicates our understanding of how and to what extent CYP450 would be affected, and further work is needed in this area. Nevertheless, it is suggested that there would be clearance-associated pharmacokinetic interactions with antivirals and other drugs that are administered in this situation[57-61]. Common drugs affected by alterations in the CYP450 pathway could include Remdesivir, which is extensively metabolized by CYP450s, particularly CYP3A4, as well as Chloroquine and Colchicine which are both included in clinical trials researching COVID-19 treatment regimes[57-61].
Pathogenesis of HRS in COVID-19 induced liver injury
The multifactorial components of COVID-19 induced liver injury leads to the development of splanchnic vasodilation, with or without portal hypertension[62]. Splanchnic vasodilation is recognized as one of the major causative factors of HRS and occurs as the result of a plethora of vasodilatory responses[63]. With increased severities of hepatic damage, there is increased production of nitrous oxide in the splanchnic bed with reduced production in liver sinusoidal cells, which lead to increased portal gradient pressures[64]. Greater levels of other vasodilating peptides such as calcitonin generelated peptide and adrenomedullin are also observed, as a result of increased production and reduced hepatic clearance[65].
Splanchnic vasodilation creates a state of hypovolemia in the central circulation, as splanchnic vasodilation combined with restricted portal blood flow causes blood to pool in the splanchnic circulation[66,67]. The body’s physiological response to central hypovolemia will eventually lead to dysregulation of blood pressure, due to abnormalities in the baroreflex and cardiovascular responses to angiotensin II, norepinephrine, and vasopressin[68]. Central circulatory dysfunction can cause cardiomyopathy affecting both systolic and diastolic heart function. There will be electrophysiological alterations, which includes QT interval prolongation and electromechanical dyssynchrony[63,64]. The ability of the heart to respond to inotropic and chronotropic stimuli is reduced. Because of decreased systemic vascular resistance, cardiac output (in absolute terms) would be maintained at a high level initially[63,64]. However, the impaired cardiac function due to the aforementioned cardiac physiological changes will become clinically apparent with normalization of systemic vascular resistance and when there are further stress stimuluses[62-64].
Ultimately, renal perfusion pressure and blood flow to the kidneys will be reduced as a result of the various mechanisms which cause central circulatory dysfunction[68,69]. Overactivation of the sympathetic system as a homeostatic response could initially increase the kidney’s reliance on blood pressure levels to maintain its perfusion[68]. Reduced blood flow to the kidneys would lead to more active stimulation of both β-adrenergic and subsequently α-adrenergic receptors, which results in afferent and efferent arteriole constriction[69]. The pathophysiological process of kidney damage is exacerbated by an inability of the liver in HRS to degrade renin, which will lead to persistent stimulation of the renin-angiotensin-aldosterone axis[70].
There are other theories of how HRS may manifest following severe liver injury from COVID-19 infection. One relates to the impact of reduced hepatic blood flow to kidneysviathe hepatorenal reflex and exacerbated further by cytokine-induced vasoconstriction, which alters kidney hemodynamics[62,71]. Animal studies have highlighted that an acute increase in portal vein pressure results in increased renal nerve activity, although this phenomenon does not occur when the liver is denervated[71]. In studies assessing empirical treatment of HRS, a lumbar sympathetic block has been shown to improve kidney function [sodium excretion, blood flow and estimated glomerular filtration rate (eGFR) were shown to be improved][72]. This may explain why medical procedures such as transjugular intrahepatic portosystemic shunt (TIPS) improve HRS in many patientsviareduction of portal pressure gradients.Another theory explaining the development of HRS in this context may relate to the direct effects of intra-abdominal ascitic pressure, in patients with underlying decompensated CLD where the ascites may be exacerbated following COVID-19 infection[73]. Increased intra-abdominal ascitic pressure can lead to venous congestion and stimulation of the renin-angiotensin-aldosterone system, resulting in further kidney function decline and histopathological changes[73].
CLlNlCAL ASSESSMENT AND MANAGEMENT STRATEGlES lN COVlD-19 lNDUCED HRS
Key components of clinical assessment and management in COVID-19 induced HRS are summarized in Table 1. Clinical assessment of patients presenting with COVID-19 induced HRS should encompass a holistic understanding of an individual’s medical history. This should be followed by physical examination to elicit specific signs, before urine, serum and imaging investigations are conducted, with these investigations forming the crux of the diagnostic criteria. Management strategies in this scenario should focus on achieving spontaneous recovery of liver function and resolution of HRSviamedical management and only if this fails, then to consider the potential for liver transplantation.
Clinical assessment of COVID-19 induced HRS
Our current understanding of the typical signs and symptoms which appear with COVID-19 induced liver injury and HRS remains premature, and there are likely non-specific presentations in most instances. It is reasonable to suggest patients who develop acute liver injury following a positive COVID-19 diagnosis would likely have a COVID-19 infection severe enough to present as such[74].From a thoroughly taken medical history, clinicians should aim to determine the likely course of COVID-19 infection and rule out other differentials more likely to explain the development of acute liver injury/fulminant liver failure[57]. There should be close observation for systemic (i.e., septic symptoms, monitor hemodynamic stability as likely to have low mean arterial pressure) as well as respiratory-specific signs and symptoms[57]. It has been reported from prospective studies conducted in China that risks of severe liver injury is greater in patients who develop gastrointestinal symptoms such as diarrhea, nausea and vomiting, anorexia and abdominal pain (OR 2.71, 95%CI 1.52-4.83,P< 0.05)following acute COVID-19 infection[75]. Given patients with underlying CLD such as cirrhosis are more likely to develop HRS regardless of COVID-19 status, classical features of CLD/decompensated liver disease including jaundice, altered mental status, malnutrition, and ascites (ascites resistant to the use of diuretic medications is characteristic of type 2 HRS) should be meticulously monitored[76]. Whilst there is relative clarity regarding hepatic signs and symptoms in HRS, the same cannot be said for renalspecific signs and symptoms. Both oliguria and normal levels of urine output have been observed for patients diagnosed with HRS[62]. Due to inability in establishing definitive renal symptoms in HRS,there is wide opinion that HRS should be diagnosed mainly on the basis of laboratory results rather than symptomatic presentation.
The utilization of diagnostic tests should initially confirm COVID-19 statusviaa real-time reversetranscriptase-polymerase chain reaction (rRT-PCR) test and determine the severity of disease through chest imaging (i.e., chest X-ray or computed tomography)[77]. Serum tests evaluating the systemic inflammatory state (i.e., full blood cell count, C-reactive protein, interleukin-6 Levels) should follow alongside tests to identify the presence of liver pathology, typically indicated by the rise in serum ALT,AST, total bilirubin, gamma-glutamyl transferase (GGT), alkaline phosphatase (ALP) levels and reduction in serum albumin[78]. Recent observational data reported the pooled prevalence of the elevated liver enzymes ALT, AST, and total bilirubin in COVID-19 positive patients to be 18% (95%CI 13%-25%), 21% (95%CI 14%-29%), and 6%(95%CI 3%-11%), respectively[79]. These serum tests may prove to be a strong marker of poor prognosis, as a fatal outcome with COVID-19 induced liver disease is estimated to be between 58% and 78%[80]. The presence of hypoalbuminemia also signifies a more severe disease process with poorer prognosis[81,82]. Liver ultrasound could also be a useful front-line diagnostic imaging tool to detect the presence of any acutely developed liver lesions.
Investigations to ascertain kidney function in confirming a HRS diagnosis should encompass urinalysis, a serum urea & electrolytes screen and other renal panel testing to rule out differential diagnoses[83-86]. Important results to look out for in urinalysis is concentrated urine with low urine sodium (< 10 mmol/L) where there is usually no proteinuria or hematuria[85,86]. There would be absence or few granular (hyaline or muddy-brown) casts identified in urine microscopy, in contrast to acute tubular necrosis (ATN) which is a known renal complication of cirrhosis[84]. ATN in the context of liver injury most likely occurs as a result of exposure to toxic medications or the development of decreased blood pressure, and proximal tubular cells are unable to reabsorb sodium from urine[62,84,85]. Because of this, urinary sodium levels in ATN would be expected to be higher than that of HRS[84].It is expected that there is marked reduction in eGFR with HRS, with no improvements in kidney function despite treatment with intravenous fluids (kidney function improvements are observed in most other causes of pre-renal kidney failure following intravenous fluid administration with reduction in serum creatinine and increased sodium excretion) due to the intra-renal vasoconstricted state[87]. Serum sodium concentration would be low due to retention of fluid together with sodium leading to dilutional hyponaetremia[88]. Plasma renin activity would be elevated considering the metabolic changes in HRS[89]. Kidney ultrasound would rule out obstruction of the kidney outflow tract.

Table 1 Key components of clinical assessment and management in COVlD-19 induced hepatorenal syndrome
Management strategies in COVID-19 induced HRS
The mainstay of treatment in HRS from either acute liver failure or cirrhosis, whether the cause is COVID-19 related or not, is usually medical management to aim for gradual liver recovery and resolution of HRS, with supportive dialysis or haemofiltration if required. Liver transplantation would not be indicated if recovery of liver function and resolution of HRS is achieved solely through medical management, with this usually resulting in the best outcome for patients. A key priority is to limit druginduced hepato- and nephrotoxicity through a dose-dependent adjustment approach of managing immunosuppressive medications, antivirals and other COVID-19 treatment regimes if and when COVID-19 treatment is indicated[57].
The combination of extracorporeal membrane support and dialysis in HRS demonstrated significant effects in removing toxins from the circulation, including systemic inflammatory molecules generated from COVID-19 infection[83,90-92]. There has been greater use of molecular adsorbents recirculation systems in HRS as both a supportive treatment option or as a bridging therapy to liver transplantation if indicated, though wider work is needed to improve accessibility with this technology still being relatively novel[93]. Close haemodynamic monitoring during hemodialysis is recommended, given concerns that this may further deteriorate blood pressure stability in HRS, increasing the risk of mortality[94]. TIPS involves decompressing the high pressures in the portal circulation by placing a small stent between a portal and hepatic vein,viaplacement of a radiologically-guided catheter passed into the hepatic vein either through the internal jugular vein or the femoral vein[95]. This will theoretically reduce portal vein pressure, which as discussed in the previous section is a key factor in the hemodynamic process leading up to HRS[17,18,62,95]. Previous studies in patients with cirrhosis largely noted improvements in kidney function when TIPS is performed, particularly as a bridging treatment if liver transplantation might be indicated[96].
Intravenous albumin can expand plasma volume, and provide other benefits in the form of its immunological, antioxidant, endothelial protective functions. Combining intravenous albumin with other medical and/or procedural treatments displayed better outcomes compared to administering intravenous albumin alone[97]. Other pharmacological options which have demonstrated efficacy across all forms of HRS may include the combined use of Midodrine, an α agonist with somatostatin analogues such as Octreotide[98,99]. In Europe, Terlispressin and albumin are recommended in the best practice guidelines[100]. These drugs regulate blood vessel tone in the gastrointestinal tract, and also systemic vasoconstrictors which inhibit splanchnic vasodilation[97]. Interestingly, these drugs were only found to be effective when used in combination and not when independently prescribed[100]. There is preliminary data that other vasopressin analogues (e.g., Ornipressin), Pentoxifylline, Acetylcysteine and Misoprostol amongst other treatments are potentially useful treatments in HRS, but this will require further study[101-103].
Liver transplantation would be the ideal treatment in HRS, if renal function cannot be corrected with medical management and hepatic recovery is unlikely with conservative management alone. These situations mostly occur in patients with CLD, where the medical management options aforementioned serve as bridging therapies towards transplantation[17,18,20,62]. The optimal strategy would usually observe effects from treatment of the underlying cause of HRS first before planning for liver transplantation. During the COVID-19 pandemic, most hepatology societies advised the deferral of liver transplantation in stabilized patients[104]. There has been continuous debate throughout the pandemic on how to optimize the procedures of liver transplantation for patients with COVID-19 positive status,such as those with active COVID-19 infection inducing HRS[105]. Currently, there is only one reported case of liver transplantation in HRS with COVID-19 infection, performed 28 d after hospital admission[23]. Individuals with HRS who receive liver transplantation almost universally achieve recovery in kidney function[91]. Previous studies have demonstrated that survival rates at 3-year follow-up for liver transplant recipients in HRS are comparable to liver transplant recipients for other causes of liver disease[106]. Although differences in long-term outcomes are significant between patients who receive liver transplantation and those who do not, acute mortality rates after liver transplantation were found to be up to 25% in the first month[107]. Patients who present with further decline in liver and kidney function following liver transplantation are at higher risk[17]. Kidney function decline following liver transplantation in HRS is usually transient and most likely attributed to drug-induced nephrotoxicity,specifically the introduction of immunosuppressants such as Tacrolimus and Cyclosporine which are known to affect kidney function[17,105].
CONCLUSlON
There is increased attention towards the extra-respiratory manifestations of COVID-19 as the pandemic continues to affect billions of lives. Hepatic consequences of COVID-19 infection are now recognized as an important complication of COVID-19. The development of HRS following COVID-19 induced liver injury suggests severe and perhaps life-threatening disease, particularly for individuals with multimorbidities including pre-existing CLD. The prognosis of HRS is largely dependent on whether liver transplantation would be viable and accessible for the patient. Confounding effects of drug-induced hepato- and nephrotoxicity in exacerbating the systemic damage from COVID-19 induced HRS should always be considered and avoided if possible. A greater understanding of the multi-faceted pathophysiological mechanisms which result in HRS following acute COVID-19 infection is important to guide clinical decisions in a timely manner for the optimization of patient outcomes.
FOOTNOTES
Author contributions:Wu HHL performed the majority of the writing, prepared the figures and tables; Athwal VS,Kalra PA, and Chinnadurai R provided review of the draft versions of the paper prior to submission of the final version; Wu HHL and Chinnadurai R designed the outline and coordinated the writing of the paper.
Conflict-of-interest statement:The authors report no conflicts of interest associated with the work presented in this manuscript.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:Australia
ORClD number:Henry H L Wu 0000-0002-4561-0844; Varinder S Athwal 0000-0002-1684-721X; Philip A Kalra 0000-0001-7652-1572; Rajkumar Chinnadurai 0000-0003-3973-6595.
S-Editor:Chen YL
L-Editor:A
P-Editor:Chen YL
 World Journal of Gastroenterology2022年39期
World Journal of Gastroenterology2022年39期
- World Journal of Gastroenterology的其它文章
- Interplay between metabolic dysfunction-associated fatty liver disease and chronic kidney disease:Epidemiology, pathophysiologic mechanisms, and treatment considerations
- Pitfalls and promises of bile duct alternatives: A narrative review
- SARS-CoV-2-induced liver injury: A review article on the high-risk populations, manifestations,mechanisms, pathological changes, management, and outcomes
- Immune checkpoint inhibitor-mediated colitis is associated with cancer overall survival
- Serum metabolic profiling of targeted bile acids reveals potentially novel biomarkers for primary biliary cholangitis and autoimmune hepatitis
- Receptor of advanced glycation end-products axis and gallbladder cancer: A forgotten connection that we should reconsider