
亲水色谱-质谱联用方法分析水中肼、甲基肼和偏二甲肼
2022-10-22 08:33徐林楠贾冬雪
含能材料 2022年10期
徐林楠,张 旭,赵 坦,贾冬雪,李 俊
(北京航天试验技术研究所,北京 100074)
0 引言
以肼(hydrazine,Hy)、甲基肼(monomethylhydrazine,MMH)及偏二甲肼(unsymmetrical dimethylhydrazine,UDMH)为代表的部分肼类化合物具有强还原性、高活性和高能量密度,是航天液体推进剂中最为常用的一类燃料[1-2]。然而,肼类化合物具有较高的毒性,可通过多种暴露途径对人体健康产生危害[3-4]。因此,国家相关卫生标准对肼类化合物的含量作出了明确的规定,例如航天推进剂水污染物中肼、甲基肼和偏二甲肼的最高允许排放浓度分别为0.1,0.2,0.5 mg·L-1[5]。
水样中肼类化合物的含量常用分光光度法进行分析[6-7]。由于肼类化合物自身不具有紫外/可见光吸收,因而对肼类化合物的分光光度分析需要借助于肼类化合物与特定显色试剂之间的反应来实现。在实际应用中,上述基于显色反应的肼类化合物分光光度分析方法存在很多的问题[8-9]:首先,方法选择性较差,由于肼类化合物性质的相似性,在分光光度法测定中3 种肼类化合物之间存在着严重的相互干扰,使得此类方法在同时含有多种肼类化合物的水样分析中准确性较差。其次,方法分析时间较长,肼类化合物与显色剂之间的反应需要较长的反应时间以保证反应完全进行,影响方法的分析效率。最后,3 种肼类在分光光度测定中需使用不同的反应试剂及反应条件,使得分光光度法无法同时对水样中所含的多种肼类化合物进行同时测定。
高效液相色谱法具有分离机理多样、选择性好、适用范围宽、分析速度快等特点,在水中肼类化合物分析方法开发中有着良好的应用前景。反相色谱是高效液相色谱最为常用的分离模式,但强极性肼类化合物在反相色谱中无法实现有效分离,仍需预先对肼类化合物进行衍生化处理[10-11]。相比之下,亲水色谱则提供了一种更为理想的选择[12-13]。一方面,亲水色谱能够为极性小分子化合物及离子化合物提供良好的分离能力,可直接应用于肼类化合物的分离分析,而无需在分离前对样品进行衍生化处理。另一方面,亲水色谱的分离流动相可使用含挥发性有机溶剂和挥发性缓冲盐的弱酸性溶剂,因而亲水色谱可通过电喷雾接口实现与高灵敏度、高选择性的质谱检测器的联用。
前期工作中,研究人员对亲水色谱-质谱联用方法分析新型推进剂中硝酸羟乙基肼含量的方法进行了研究[14]。但对于水样中微量的肼、甲基肼和偏二甲肼分析而言,还存在一些需要解决的问题:(1)水样中的肼类化合物浓度低,原有的分析条件,特别是样品制备方式无法满足高灵敏度分析的要求;(2)被分析物(特别是肼)具有更高的反应活性,其与亲水色谱分析中常用的溶剂和流动相条件的相容性有待研究;(3)肼在质谱电喷雾离子源处的离子化效率未知,可能影响其分析灵敏度。
综合上述问题,本研究尝试建立水中肼类化合物的液相色谱-质谱联用分析方法,考察优化了样品配制和进样量、流动相组成、质谱测试条件,对样品中有机溶剂的种类和比例、进样量、特征质谱信号识别和选择等影响分析准确性和灵敏度的关键条件进行了研究和分析,并对所建立的分析方法进行了方法学验证。
1 实验部分
1.1 试剂及材料
乙腈(ACN)(色谱级)、异丙醇(色谱级)、甲醇(色谱级)、甲酸铵(质谱级)购自Merck。甲酸(色谱级)购自Dikma。水为超纯水(电阻率≥18.2 MΩ·cm)。无水肼、甲基肼、偏二甲肼自制。
1.2 样品制备
在针对于样品配制条件优化的实验中,将无水肼、甲基肼和偏二甲肼使用含一定比例乙腈、甲醇或异丙醇的0.1%甲酸溶液稀释至所需浓度并分析。分析实际样品时,使用4 倍于样品体积的含0.125%甲酸的异丙醇溶液稀释样品进行分析。
1.3 液相色谱-质谱分析
样品分析使用Agilent 1260 高效液相色谱仪联用6120 质谱仪完成。色谱柱为Agilent Poroshell 120 HILIC-Z,2.1 mm×150 mm,2.7 μm。流动相使用乙腈和水的混合溶剂,分别考察流动相不同乙腈比例、甲酸铵溶度和pH 下的分离效果,优化得到如下流动相条件:流动相A:乙腈/水(V乙腈/V水=10/90),10 mmol·L-1甲酸铵,pH=3.5;流动相B:乙腈/水(V乙腈/V水=90/10),10 mM 甲酸铵,pH=3.5;流动相A 和B 以VA/VB=25/75的比例进行混合。柱温30 ℃,流速0.2 mL·min-1。考察不同进样体积下的分离效果和分析灵敏度,选择进样体积为10 μL。
质谱离子源为电喷雾离子源。质谱分析在正离子模式下进行,毛细管电压4000 V。干燥气流速8 L·min-1,雾化气压力20 psi,干燥气温度300 ℃。利用扫描模式或选择离子监测(SIM)模式进行分析。扫描模式下扫描范围20~150 Da;选择离子监测模式下根据优化结果选择待测离子及碰撞诱导解离参数。
2 结果与讨论
2.1 样品配制及进样量
在使用亲水色谱-质谱联用法进行分析时,样品中溶剂的组成对被分析物在亲水色谱上的分离效果有一定的影响。在亲水色谱上,水是强洗脱溶剂,当样品溶剂中仅含水而不含有机溶剂时,较大的进样体积可能会引起被分析物色谱峰峰形变差,进而影响其分离效果和检测灵敏度。故对于含肼水样的分析,需要在进样前向样品中掺入一定比例的有机溶剂。
2.1.1 样品中添加的有机溶剂种类
在选择有机溶剂时需满足如下条件:1)有机溶剂与水混溶以保证样品的均匀性;2)有机溶剂不与肼类化合物发生化学反应以避免对分析过程造成干扰;3)有机溶剂应在亲水色谱上具有较弱的洗脱能力以降低样品溶剂整体的洗脱强度。
分别尝试使用乙腈、甲醇和异丙醇作为样品中掺入的有机溶剂。乙腈是亲水色谱流动相中最为常用的有机洗脱溶剂。相比于醇类有机溶剂,乙腈的洗脱能力更弱,用于样品溶剂配制时可很大程度上降低样品溶剂的洗脱强度。然而在本研究中,当使用乙腈作为有机溶剂时,除检测到肼的信号,还检测到保留时间与肼不同的另一化合物的信号。该化合物质谱信号的质荷比为74,依保留时间判断其极性弱于肼,依同位素分布判断其分子式为C2N3H7,推测为肼与乙腈的加成反应产物乙亚胺酰肼(ethanimidic hydrazide,EIH)(Scheme 1)。向含肼样品中加入乙腈,分别在加入乙腈后0、0.5、1、3、6、24 h 进样分析,记录肼及乙亚胺酰肼不同时间的色谱图,如图1 所示。可见加入乙腈后,随肼和乙腈加成反应的进行,所检测到的肼的峰面积迅速下降。若使用乙腈作为添加的有机溶剂,则肼含量的测试会受到样品配制时间的干扰。因此,在进行肼类化合物分析时,乙腈不适合作为添加至样品中的有机溶剂。
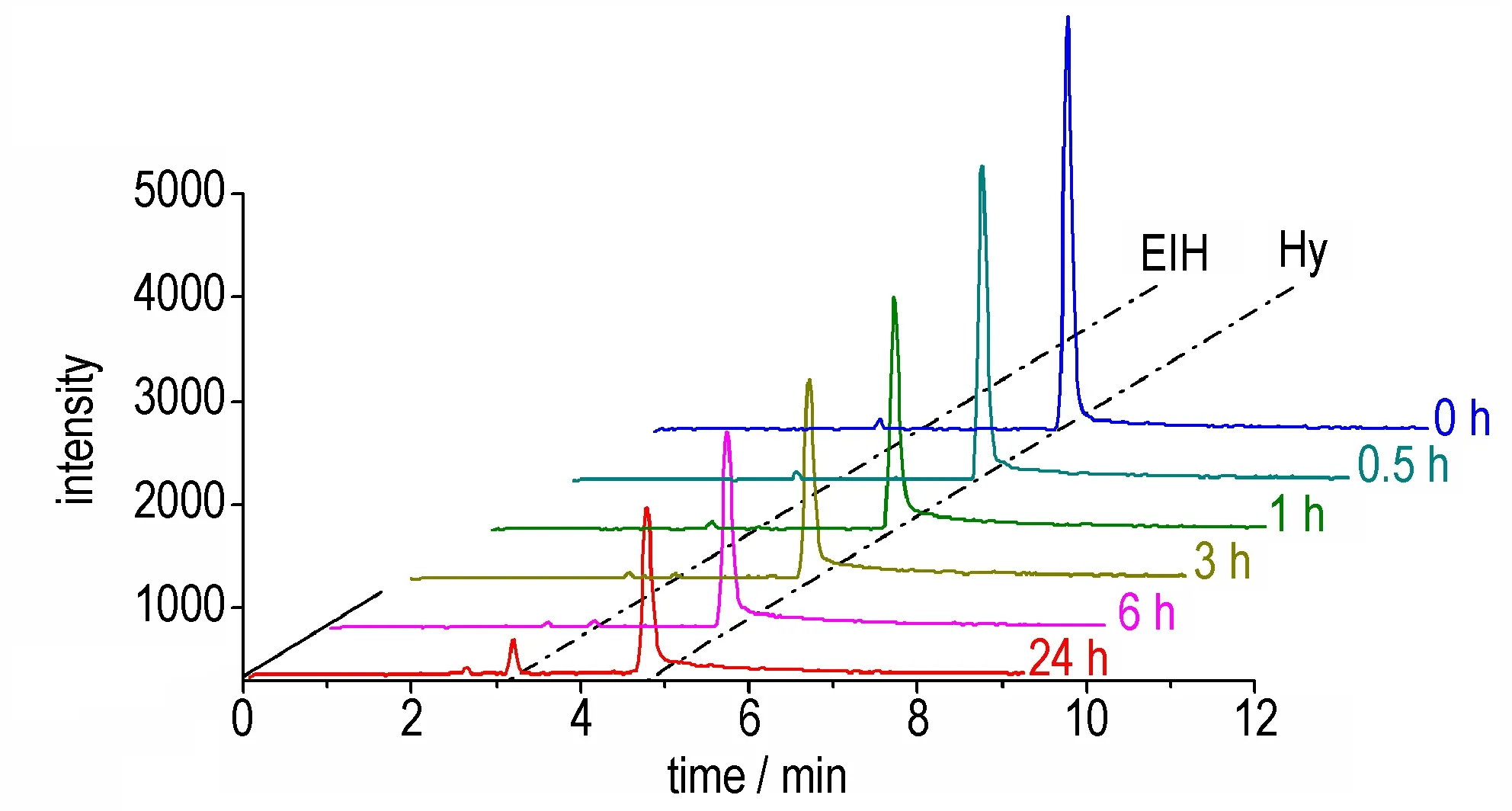
图1 不同反应时间下肼及乙亚胺酰肼的提取离子流图Fig.1 Extracted ion chromatograms of hydrazine and ethanimidic hydrazide under different reaction time

Scheme 1 Speculated product of reaction between hydrazine and acetonitrile
甲醇、异丙醇等醇类溶剂不会与肼类被分析物发生反应,因而可作为样品溶剂使用。在亲水色谱上,异丙醇比甲醇极性低,相比下具有更弱的洗脱强度,理论上更适合作为样品中添加的有机溶剂使用。向含3 种肼类化合物的样品中分别添加70%甲醇和70%异丙醇并进样4 μL 进行分析,经色谱分离后,3 种肼类化合物的柱效如表1 所示。使用甲醇时,肼的色谱峰出现了分叉,并且甲基肼和偏二甲肼的柱效均低于使用异丙醇时的结果。相比之下,使用异丙醇作为样品添加的有机溶剂能够获得更高的柱效,也即意味着更好的分离效果和更高的检测灵敏度。因此,选择异丙醇作为样品溶剂中添加的有机溶剂。
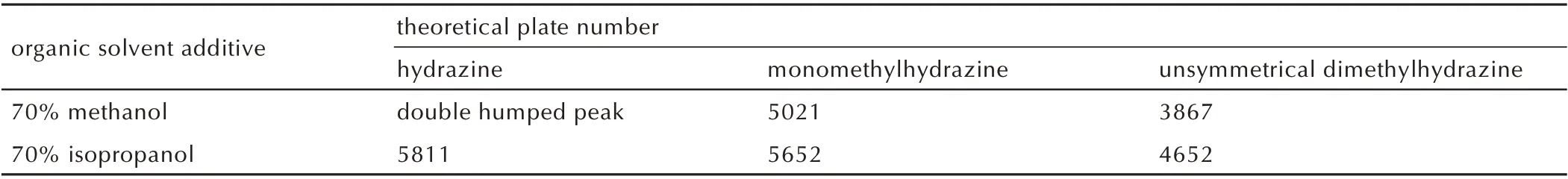
表1 分别使用甲醇和异丙醇作为样品添加有机溶剂时的柱效Table 1 Column efficiency with methanol / isopropanol as organic solvent additive in samples
2.1.2 样品中异丙醇添加比例及进样量
向样品中添加高比例异丙醇有助于分离效果的改善,但同时也会引起样品过度稀释,影响分析方法的灵敏度。与之类似,样品进样量也同样会对分析灵敏度及分离效果产生作用相反的影响:大进样量可以提高分析过程中实际进入分析系统的被分析物含量,从而提高方法的灵敏度。但过大的进样体积会导致被分析物色谱峰出现展宽、拖尾、分叉等不利于化合物分离的情况。对异丙醇的添加比例和进样量进行了考察优化。为消除不同异丙醇添加比例对进样量的影响,对不同异丙醇添加比例的样品使用不同的进样量,以保证进行比较的实际进样体积(即实际进入分析系统的不含异丙醇的样品溶液体积)一致。分别向样品溶剂中添加50%、70%和80%异丙醇,并考察实际进样体积为1.5、2、3 μL 时肼的分离效果(相比于甲基肼和偏二甲肼,肼在亲水色谱中更容易发生峰分叉的现象,影响其分离和定量检测),结果如表2 所示。
由表2 可见,当实际进样体积为3 μL 时,肼的色谱峰均出现分叉的情况,无法用于肼含量的分析。当实际样品进样量为2 μL 时,向样品中添加70%异丙醇(稀释后样品进样量为6.67 μL)或添加80%异丙醇(稀释后样品进样量为10 μL)时肼均为单谱峰,其中添加80%异丙醇时肼的色谱峰具有更高的柱效。相比于1.5 μL 的实际进样体积,采用2 μL 实际进样体积可在保证分离效果的基础上获得更高的进样量和分析灵敏度。综上,选择80%作为异丙醇添加比例,并选择10 μL 作为稀释后样品的进样体积。

表2 不同异丙醇添加比例和不同进样量条件下肼的柱效分析Table 2 Column efficiency with different isopropanol percentage and different injection volume
2.2 流动相组成
流动相组成是影响肼类化合物在亲水色谱上分离效果的决定性因素,同时也会对被分析物在电喷雾质谱上的检测灵敏度产生显著的影响。亲水色谱的流动相通常由水、有机溶剂、挥发性小分子有机弱酸及其盐组成,其中有机溶剂通常使用洗脱强度相对较弱的乙腈。尽管前述实验中观察到肼会与乙腈发生反应,但由于肼在色谱中的保留时间较短(~4 min),且保留时间具有高度的重现性,因此肼与流动相中乙腈的反应对肼含量的影响并不显著,且可通过外标法进行校准,故本方法中使用乙腈作为流动相中的有机溶剂。
2.2.1 流动相中乙腈的比例
流动相中乙腈的比例影响到被分析物在亲水色谱上的保留时间。提高乙腈的比例通常能够带来更好的分离效果,但过高的乙腈比例也会延长被分析物的保留时间,从而影响方法的分析效率。
本研究对流动相中乙腈的比例进行了考察优化,以在保证色谱完全分离和质谱高灵敏检测的基础上,缩短样品分析所需的时间。3 种肼类化合物保留时间tR及分离度R随流动相中乙腈含量的变化分别如图2a和图2b 所示。可见随乙腈比例的提高,3 种肼类化合物的保留时间和分离度均逐渐增大。在所试验最低的流动相乙腈比例下,3 种组分在谱图上的分离度即均满足R≥1.5 的基线分离要求。
继续考察流动相不同乙腈比例对检测灵敏度的影响。3 种肼类化合物在色谱图上的峰高随流动相中乙腈含量的变化如图2c 所示。可见偏二甲肼和甲基肼在乙腈含量70%时具有最高的检测灵敏度,而肼的检测灵敏度在试验范围内随流动相乙腈比例的提高而下降。综合考虑分离效果、检测灵敏度、单次分析时间等因素,选择70%作为流动相中乙腈的比例。在该乙腈比例下,3 种被测组分能够实现完全分离,单次分析时间较短,且3 种肼类化合物均能获得较高的检测灵敏度。
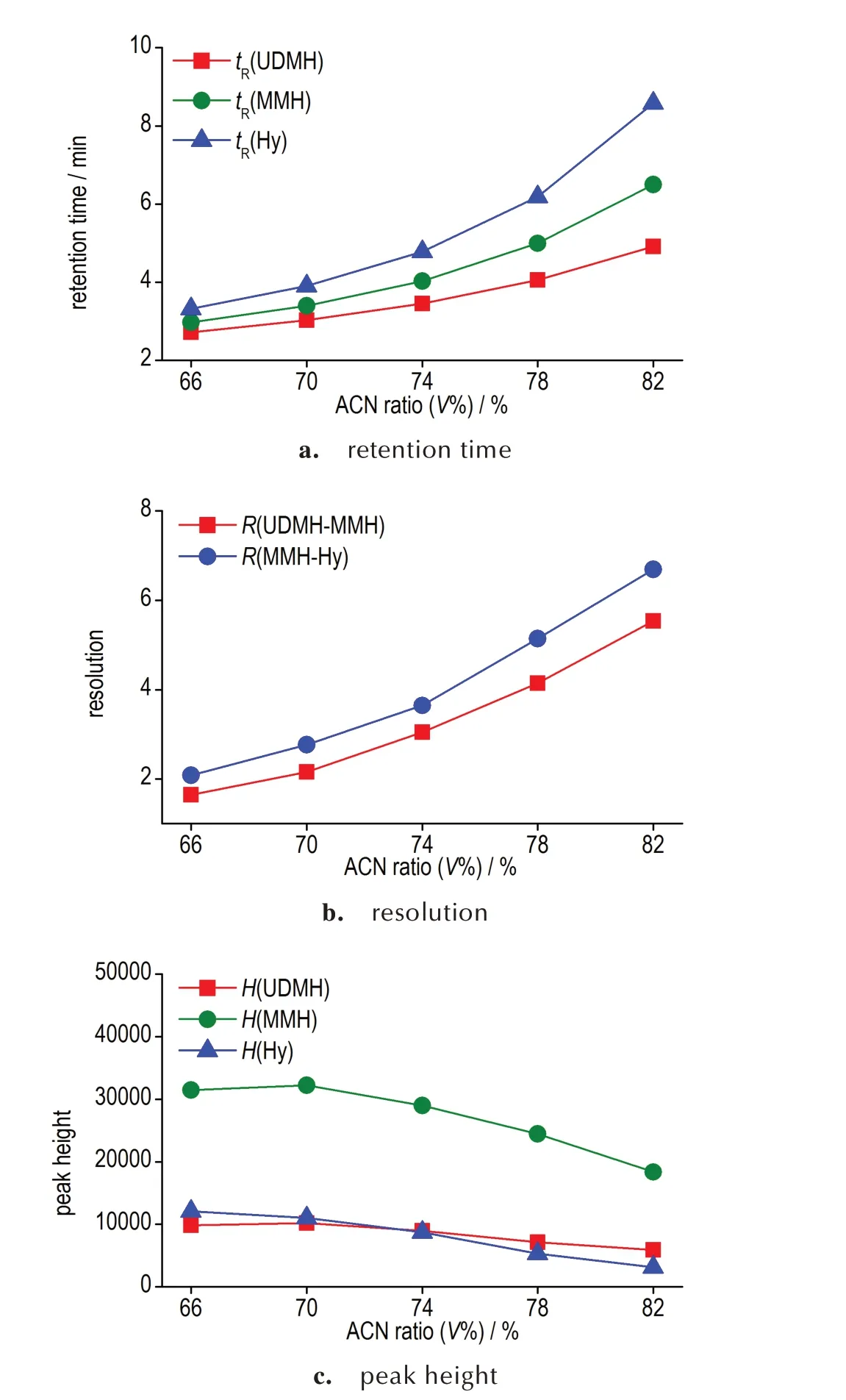
图2 不同流动相乙腈比例下3 种被分析物的分离效果Fig.2 Separation efficiency of three analytes with different ACN ratio in mobile phase
2.2.2 流动相pH
作为一类弱碱性化合物,肼类化合物的电离形式会随溶液pH变化而改变。同时,亲水相互作用色谱固定相表面的电荷分布情况也会受溶液pH影响。因此,流动相pH条件会对肼类化合物的分离效果产生影响。
由于肼类化合物结合氢离子获得正电荷后才能够被质谱检测到,因此需要流动相保持在弱酸性条件下。本研究中使用甲酸-甲酸铵缓冲对提供流动相所需的弱酸性条件。通过调节流动相中甲酸和甲酸铵的相对比例,可在一定范围内为流动相提供不同的pH 条件。在pH为2.5~4.5范围内,考察了不同pH下3种肼类化合物分离效果,其结果如图3a 和3b 所示。可见在所试验的范围内流动相pH对分离效果的影响并不十分显著。
继续考察流动相pH 对检测灵敏度的影响。由图3c可见,在pH 2.5~3.5 的范围内,3 种肼类化合物的峰高变化不显著;当pH>3.5 时,3 种肼类化合物的峰高则呈现随pH 升高缓慢下降的趋势。考虑到在2.5~3.5 的范围内pH 对3 种肼类化合物的检测灵敏度没有显著影响,且较高的流动相pH 能够减少流动相配制过程中高纯甲酸的用量,故选择3.5 作为流动相pH。

图3 不同流动相pH 下3 种被分析物的分离效果Fig.3 Separation efficiency of three analytes with different mobile phase pH
2.2.3 流动相中甲酸铵的浓度
在亲水色谱中,向流动相中掺入一定浓度的缓冲盐有助于改善被分析物的峰形,提高检测灵敏度。但流动相中过高的缓冲盐浓度也会影响被分析物在质谱离子源处的离子化效率,降低被分析物的检测灵敏度。
本研究考察了流动相中甲酸铵浓度对分离效果和检测灵敏度的影响,分别在甲酸铵浓度为5、10、15、20、25 mmol·L-1的流动相中对含有3 种肼类化合物的同一样品进行分析。3 种肼类化合物的保留时间及分离度随甲酸铵浓度的变化关系如图4a 和图4b 所示。值得注意的是,当甲酸铵浓度为5 mM 时,肼的色谱峰出现的分叉的情况,因此无法确定其保留时间。这也表明过低的缓冲盐浓度并不适合于肼的分析。在10~25 mM 的范围内,缓冲盐浓度对保留时间无显著影响。随缓冲盐浓度的升高,偏二甲肼-甲基肼的色谱分离度缓慢升高,而甲基肼-肼的色谱分离度则呈现缓慢下降的趋势。

图4 不同流动相甲酸铵浓度下3 种被分析物的分离效果Fig.4 Separation efficiency of three analytes with different ammonium formate concentration in mobile phase
相比于分离效果,甲酸铵浓度对肼类化合物的检测灵敏度的影响则十分显著。由图4c 可见,随着甲酸铵浓度的逐渐提高,3 种肼类化合物的峰高都明显下降,表明其检测灵敏度随甲酸铵浓度的提高而下降。这是由于高浓度甲酸铵的存在会干扰肼类化合物在离子源处的离子化效率,导致可被检测到的化合物绝对量减少。综合考虑分离效果和检测灵敏度,选择10 mM 作为流动相甲酸铵的浓度。
综合上述实验结果,最终确定的流动相条件为70%乙腈、10 mM 甲酸铵、pH=3.5。
2.3 质谱测试条件
2.3.1 质谱信号选取
被分析物分子结合氢离子发生质子化,是电喷雾离子源中最常见的离子化方式[15]。对于大多数被分析物,质子化被分析物的质谱信号是谱图上强度最高、最适合用于被分析物检测和定量的信号。通过对质荷比20~150 范围内的全扫描分析可知,质子化甲基肼([MMH+H]+,m/z=47)和质子化偏二甲肼([UDMH+H]+,m/z=61)分别为甲基肼和偏二甲肼强度最高的质谱信号,故选择质荷比47 和61 分别作为甲基肼和偏二甲肼的质谱检测信号。
但对于肼,实验发现肼质子化形成的质荷比为33的信号在质谱上的强度和灵敏度极差。相比之下,在低碰撞能条件下对肼进行分析时,则检出质荷比为74的高强度信号(如图5 所示)。通过对分析系统中所存在化合物及对同位素峰相对丰度的分析确定,该信号来自于肼-乙腈-氢离子所形成的非共价加合物([Hy+ACN+H]+,m/z=74)。与前述3.1.1 中所描述的肼与乙腈发生加成反应生成乙亚胺酰肼不同的是,在质谱上所检测到的加合物中肼与乙腈并未发生化学反应,仅在质谱离子源处通过非共价相互作用结合而被一同检测出来。由图5 中两质荷比的离子流图可知,该非共价加合物的质谱信号与质子化肼的质谱信号具有相同的保留时间(~4.5 min),表明二者为同一物质(肼)在质谱中的不同存在形式。由于该加合物信号能够提供更高的肼检测灵敏度,故选择质荷比74 作为肼的检测信号。
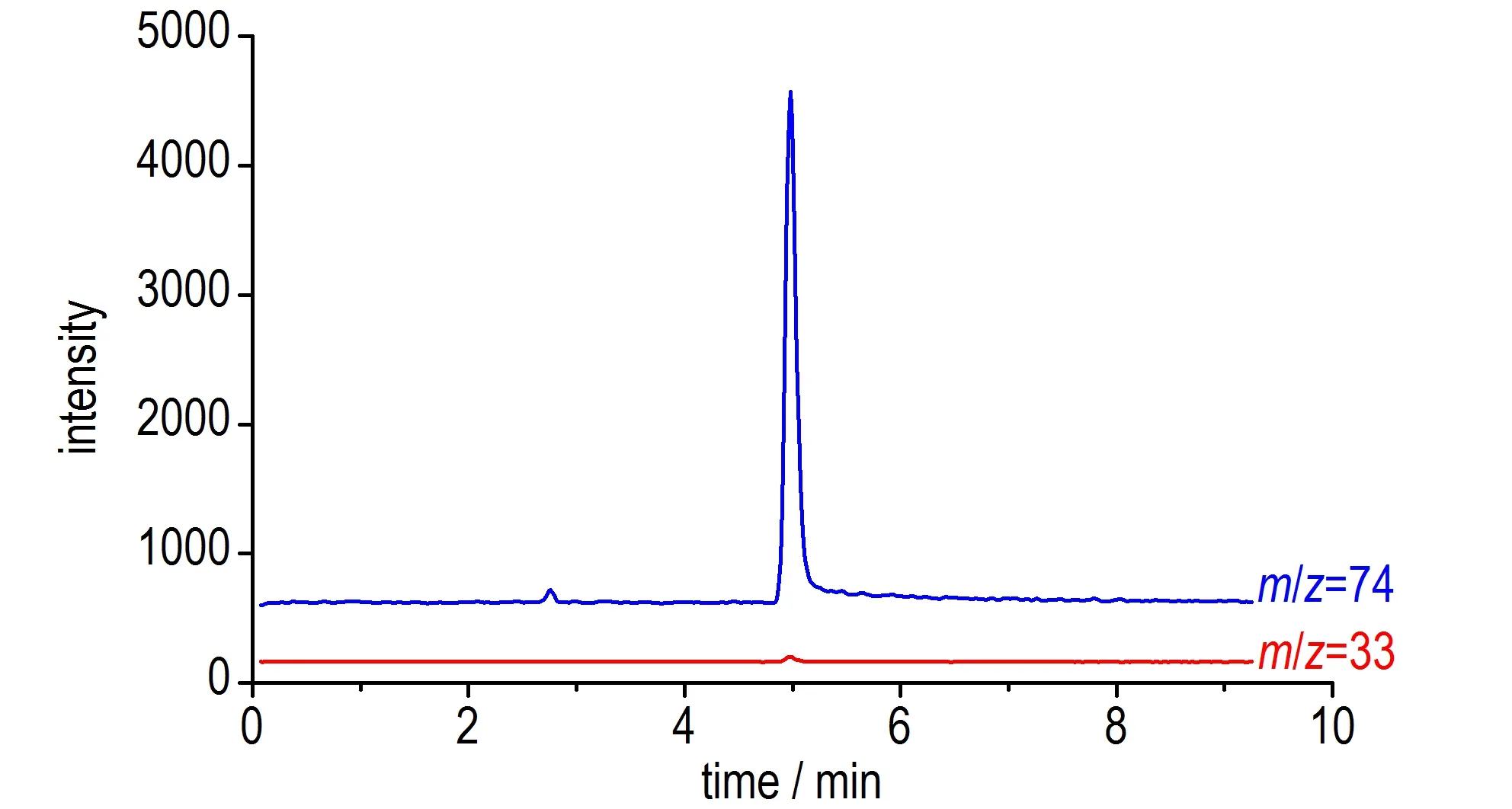
图5 肼在不同监测离子下的离子流图Fig.5 Ion chromatograms of hydrazine under different monitored ion
2.3.2 源后碰撞能
源后碰撞能也是质谱分析中的关键参数。质谱中的碰撞是利用惰性气体分子以一定的能量与进入离子源中的目标化合物发生碰撞,借助于碰撞的能量破坏目标化合物中的共价键或非共价键。碰撞过程可以有效消除溶剂分子、缓冲盐离子之间形成的各类干扰被分析物分析的加合离子,一定程度上提高被分析物的检测灵敏度。但过高的碰撞能则会导致目标化合物及其加合离子的碎裂,影响被分析物的检出。
本研究考察了源后碰撞能在0~200 eV 范围内对3 种肼类化合物质谱峰面积的影响,其结果如图6 所示。对于甲基肼和偏二甲肼,当碰撞能小于100 eV 时,随着碰撞能的提高,流动相中的离子化组分(主要是由乙腈与铵根离子形成非共价加合物离子,[ACN+NH4]+,m/z=59)逐渐减少,甲基肼和偏二甲肼的离子化效率逐渐提高,因而其质谱信号强度逐渐增大。当碰撞能大于100 eV 时,甲基肼和偏二甲肼的共价键会在高能量碰撞下碎裂,导致实际可检测到的质子化甲基肼和质子化偏二甲肼减少。根据其信号强度与源后碰撞能的变化关系,选择信号强度最高的100 eV 作为甲基肼和偏二甲肼质谱检测信号的源后碰撞能。但对于肼而言,由于其检测的信号离子为非共价加合物,该加合物更容易在碰撞过程中发生解离。当碰撞能高于50 eV时,质谱信号强度即随碰撞能的提高而下降,故选择50 eV 作为肼质谱检测信号的源后碰撞能。
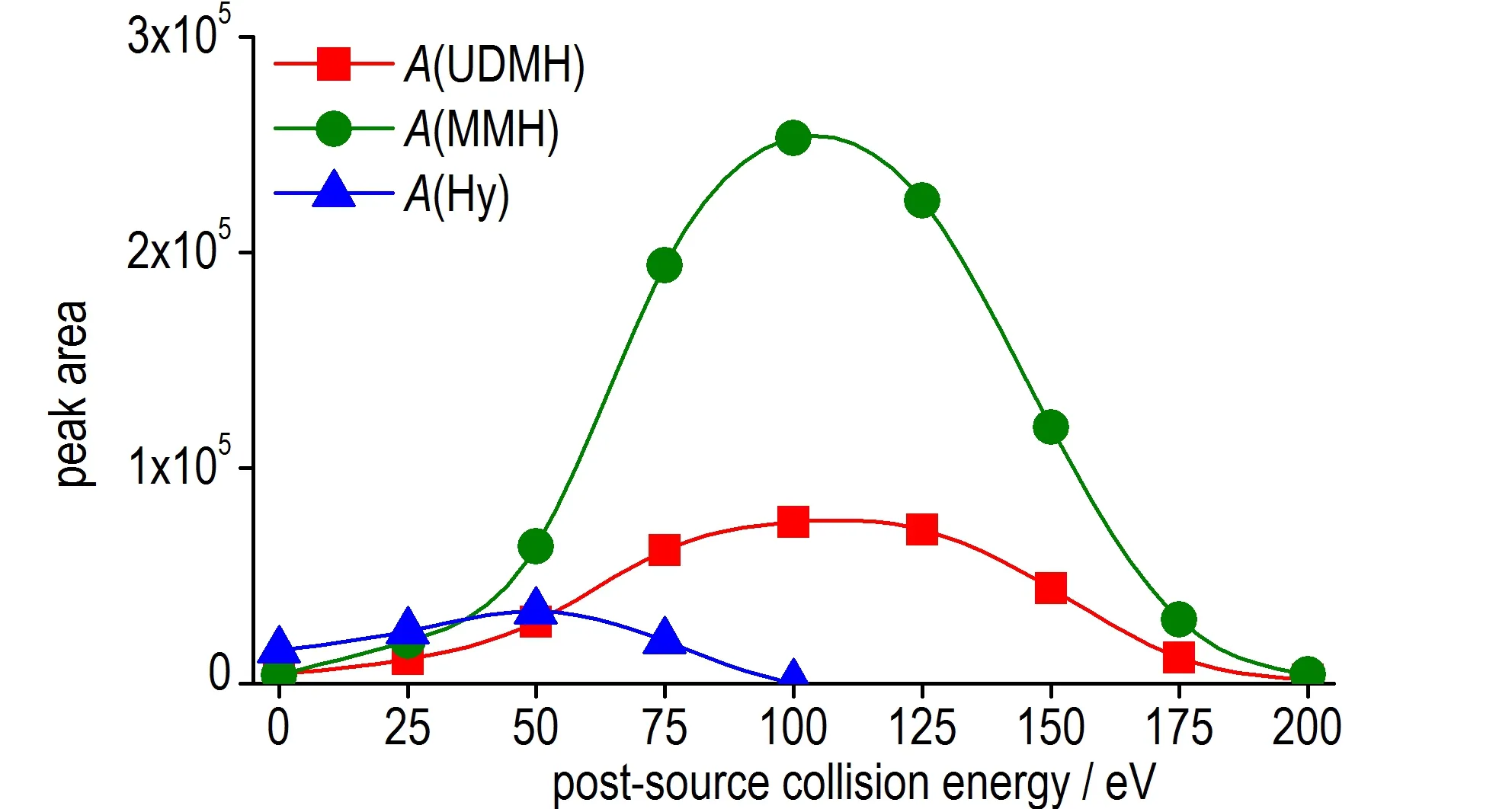
图6 3 种被分析物信号峰面积随源后碰撞能的变化关系图Fig.6 Relationship between peak area of three analytes and post-source collision energy
最终确定的质谱检测分析条件列于表3。
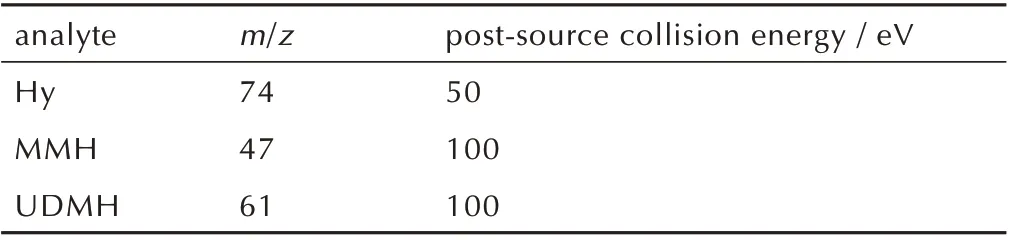
表3 3 种化合物的质谱检测条件Table 3 Mass spectrometry parameters for three analytes
2.4 线性范围和检出限
在0.01~2 mg·L-1的浓度范围(含80%异丙醇的样品浓度)内测试3 种肼类化合物峰面积与浓度的对应关系,确定其工作曲线的浓度范围及线性响应关系。工作曲线的确定采用最小二乘法,所给出的工作曲线的范围通常不小于一个数量级。为避免低浓度点在工作曲线拟合中权重过小影响低浓度范围内的样品测定准确度,采用浓度的倒数作为权重因子进行加权线性回归分析。所得3 种肼类化合物的线性范围及其线性回归方程如表4 所示。
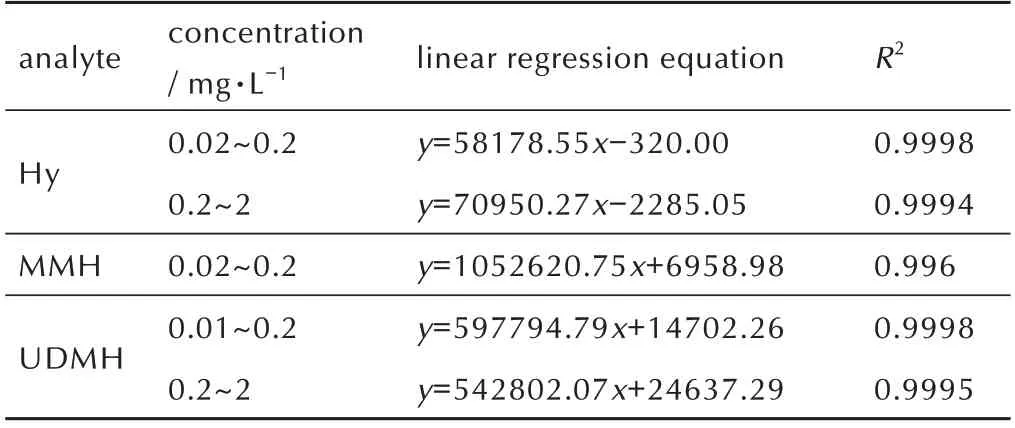
表4 3 种被分析物的线性范围Table 4 Linear range of three analytes
测试低浓度下3 种肼类化合物的信噪比,以三倍信噪比下被分析物浓度确定其检出限。对于直接用于进样分析的样品,肼、甲基肼和偏二甲肼的检出限分别为0.008、0.001、0.005 mg·L-1。考虑到样品中加入了80%异丙醇以改善分离效果,实际应用于水样分析的方法检出限分别为0.04、0.005、0.025 mg·L-1。
2.5 加标回收率
分别向纯水和含肼水样中加入3 种肼类化合物,测定加标浓度以计算加标回收率,所得结果如表5 所示。依据方法的线性范围选择加标浓度,其中肼和偏二甲肼的加标浓度介于0.1~7.5 mg·L-1,甲基肼的加标浓度介于0.25~0.75 mg·L-1,进样前均按照样品制备流程进行稀释。分别测量加标前后水样中3种肼类化合物的浓度,并计算加标被分析物的测定值和方法回收率。可见方法对于3 种肼类化合物的加标回收率介于92%~115%之间,且被分析物浓度不低于0.5 mg·L-1时,方法回收率介于96%~104%之间。该结果表明该方法对于水样中微量肼类化合物的分析具有较高的分析准确度。
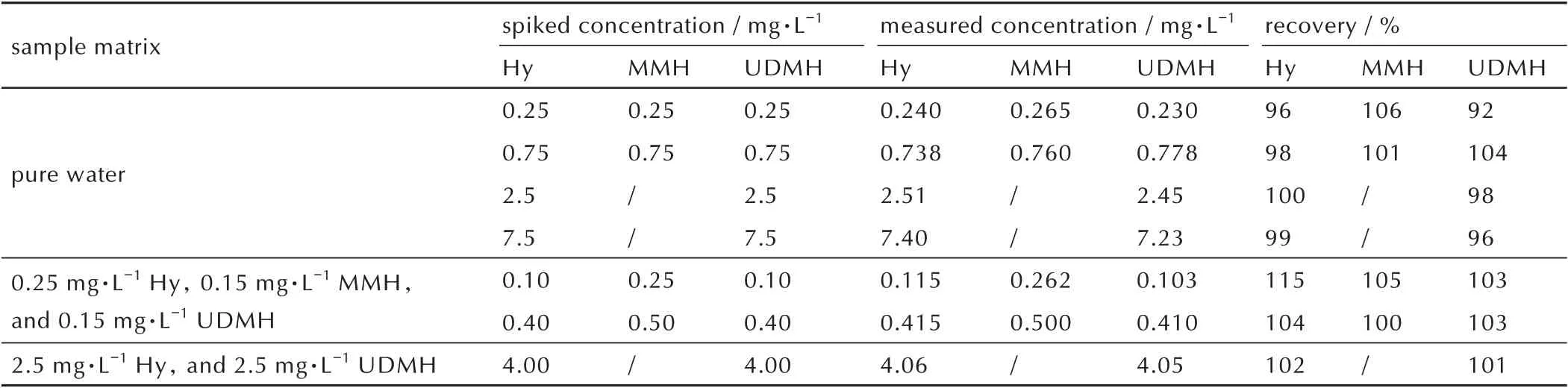
表5 加标回收率测定结果Table 5 Recovery of spiked samples
3 结论
针对水样中微量肼类化合物含量的快速分析需要,开发了基于亲水色谱-质谱联用的水中肼、甲基肼、偏二甲肼分析方法。该方法使用80%异丙醇-0.1%甲酸溶液作为样品溶剂,并进样10 μL 进行分析;以70%乙腈-10 mmol·L-1甲酸铵、pH=3.5 的混合溶剂作为流动相,使用亲水色谱柱进行分离;分别选取质子化肼-乙腈非共价加合物,质子化甲基肼、质子化偏二甲肼作为检测离子,并分别采用50、100、100 eV 源后碰撞能对肼、甲基肼和偏二甲肼进行质谱分析。所建立的分析方法在一定的浓度范围内呈现出良好的线性响应关系,可用于水样中3 种肼类化合物的定量分析;方法具有高检测灵敏度和良好的准确性,3 种肼类化合物的方法检出限均不超过0.04 mg·L-1,测定方法回收率为92%~115%,其中对于浓度不低于0.5 mg·L-1样品的加标回收率为96%~104%。
该方法统一了不同肼类化合物的分析条件,实现了对不同肼类化合物的同时分析;消除了不同肼类化合物之间的相互干扰,提高了方法在复杂样品中的分析准确度;无需进行样品衍生化等繁琐、耗时的样品预处理过程,很大程度上缩短了分析时间,提高了分析效率。目前,该方法已应用于肼类推进剂废水及水处理样品的快速高通量分析中。
猜你喜欢
分子催化(2022年1期)2022-11-02
煤化工(2022年3期)2022-07-08
中草药(2022年9期)2022-05-06
中山大学学报(自然科学版)(中英文)(2022年2期)2022-04-12
中草药(2022年3期)2022-02-14
科学与财富(2021年13期)2021-07-04
湖北农业科学(2017年24期)2018-01-27
湖北农业科学(2014年12期)2015-01-06
中小企业管理与科技·中旬刊(2014年7期)2014-09-24
中国信息化·学术版(2013年3期)2013-06-25
