
恩诺沙星新盐的制备、表征及溶解性研究
2022-10-20 05:14王凯茹毛雅楠徐瑞涛赵兴华
中国畜牧兽医 2022年10期
王凯茹,毛雅楠,徐瑞涛,赵兴华,何 欣
(河北农业大学动物医学院,保定 071000)
恩诺沙星(enrofloxacin,ENR),又名恩氟沙星,是一种动物专用的氟喹诺酮类抗菌药物,具有抗菌谱广、杀菌力强的特点[1-2],它可以选择性地抑制微生物细胞的DNA促旋酶(一种Ⅱ型拓扑异构酶)和拓扑异构酶Ⅳ,导致细菌死亡[3-4]。在临床应用中,ENR可以治疗禽类、猪、牛等的尿路、呼吸道、肠道以及皮肤和组织等感染[5];目前也广泛应用于水产养殖中,主要用于细菌性疾病和支原体感染的治疗[6]。ENR在中性溶液中溶解度较差(0.45 mg/mL)[5],其在体内溶解后从胃排空到肠内时可能发生重结晶,导致肠内容物中ENR浓度较低[7],这种特性限制了恩诺沙星制剂的开发和应用。药物成盐/共晶是指药物活性成分(active pharmaceutical ingredient,API)和共形成物按照一定的化学计量比以离子键或非共价键结合而成的晶体,若API与共形成物之间未发生质子转移形成共晶,发生质子转移则形成盐[8-9]。大量文献研究表明,盐/共晶可有效提高药物活性成分的溶解度、渗透性、稳定性及生物利用度等[10-12]。为有效提高ENR的溶解度,本研究利用晶体工程技术制备ENR新盐,并对新盐进行表征和体外溶解速率研究,以期为提高ENR生物利用度及扩大临床应用奠定基础。
1 材料与方法
1.1 材料
ENR(纯度≥98%)、2,6-二羟基苯甲酸(2,6-dihydroxybenzoic acid,2,6-DHBA,纯度≥98%)均购自上海阿拉丁生化科技股份有限公司;ENR标准品(纯度≥98%)购自北京索莱宝科技有限公司;无水乙醇(分析纯)购自福晨(天津)化学试剂有限公司。
粉末X射线衍射仪(D8 Advance)、光谱仪(Alpha FT-IR)均购自BRUKER公司;单晶X射线衍射仪(Agilent Gemini E)购自Rigaku公司;扫描电子显微镜(MERLIN Compact)购自Carl Zeiss公司;差式扫描量热仪(DCS 3)购自METTLER TOLEDO公司;热重分析仪(TGA-209 F3)购自NETZSCH公司;高效液相色谱仪、检测器、色谱柱(SunFire C18,4.6 mm×250 mm,5 μm)均购自Waters公司;单磁力搅拌器(CCLH BASIC)购自上海小聪科技有限公司。
1.2 ENR新盐的制备
称取ENR(53.91 mg,0.15 mmol)和2,6-DHBA(23.12 mg,0.15 mmol)于10 mL离心管中,加入1 mL 50%乙醇,室温下250 r/min搅拌24 h,过滤,40 ℃烘干,过100目筛备用。
1.3 ENR新盐的单晶培养
采用溶剂挥发法制备单晶:称取ENR(35.94 mg,0.1 mmol)和2,6-DHBA (15.41 mg,0.1 mmol)于10 mL离心管中,加入3 mL 50%乙醇,250 r/min搅拌12 h,之后用微孔滤膜过滤,滤液置于通风橱中静置,使缓慢生长单晶。
1.4 表征分析
1.4.1 单晶X射线衍射(SXRD) 使用单晶X射线衍射仪进行数据采集,将大小合适的单晶从溶剂中转移到单晶衍射仪的冷气流中,测试温度为110.7 K,测试光源为Mo Kα射线(λ=0.71073 Å),采集的单晶数据使用CrysAlisPRO程序进行晶胞确定和数据还原,结构用Olex2程序解析并精修,通过Mercury 4.2.0软件绘制晶体结构。
1.4.2 粉末X射线衍射(PXRD) 使用粉末X射线衍射仪进行数据采集,将少量(3~5 mg)的药物粉末平铺于样品板,测定条件为:Cu Kα (λ=1.5418 Å)光源,电压40 kV,电流40 mA,步长为0.02°/步,测试速度为0.1 s/步,样品扫描范围为3°~35°。收集后的数据使用Origin Pro 2017软件作图分析。
1.4.3 傅里叶红外光谱(FTIR) 使用光谱仪进行测定,将粉末样品分别与溴化钾混合,取适量混合粉末于玛瑙研钵中,充分研磨混匀,压片制样进行扫描测试,采集范围为400~4 000 cm-1,分辨率为4 cm-1。
1.4.4 热分析 差示扫描量热分析(DSC):使用差式扫描量热仪进行数据收集,称取3~5 mg样品粉末,置于Mettler DSC标准铝盘中,将铝盘盖扎2个小孔盖到铝盘上压紧。测定温度范围为25~350 ℃,升温速度为10 K/min,氮气流速为50 mL/min。
热失重分析(TGA):使用热重分析仪进行数据收集,取约5 mg粉末样品置于铝坩埚内,盖上铝盖,测定温度范围为25~350 ℃,升温速度为10 K/min,氮气流速为50 mL/min。
1.4.5 扫描电子显微镜(SEM)观察 使用扫描电子显微镜观察并拍摄样品的形貌照片,工作电压为20 kV,将样品用双面胶带固定在铜板上,并用金喷射,观察样品形貌。
1.5 ENR新盐的溶解速率及溶解度考察
1.5.1 高效液相色谱(HPLC)测定方法 流动相为乙腈∶0.025 mol/L磷酸水溶液(三乙胺调节pH至2.5±0.1)=18∶82(V/V),流速为1.0 mL/min,柱温为37 ℃,检测波长为278 nm,进样量为20 μL。
1.5.2 标准曲线的绘制 精密称取ENR标准品5 mg,使用0.1 mol/L NaOH溶液溶解并定容,配制成含1 mg/mL ENR的标准母液。精密量取标准母液适量,用流动相稀释得到ENR浓度为80、40、20、10、5、2.5、1.25、0.625及0.3125 μg/mL的待测溶液,使用0.22 μm过滤器过滤后采用HPLC系统进行测定。以ENR浓度为横坐标、峰面积为纵坐标进行线性回归,得到回归方程,绘制出ENR的标准曲线。
1.5.3 ENR新盐的溶解速率及溶解度测定 对过筛后的ENR和ENR-2,6-DHBA·1/2H2O进行粉末溶出测定。以pH 6.8的磷酸盐缓冲液为溶出介质,将过量的ENR和ENR-2,6-DHBA·1/2H2O分别加入到溶出介质中,设置搅拌器速度为250 r/min,温度为37 ℃,在预设的2、5、10、15、20、30、45、60、120、240、360、480 min取200 μL溶液,立即过滤同时补充等体积新鲜介质,使用流动相稀释到合适倍数后进行HPLC的测定,试验平行3次,并在溶出试验结束后收集滤渣测定PXRD。
2 结 果
2.1 SXRD分析
培养单晶的滤液在挥发1周左右长出了透明棒状的晶体,晶体学数据显示ENR-2,6-DHBA·1/2H2O盐属于三斜晶系的P-1空间群,晶胞参数为:a=13.2495(7) Å;b=13.8266(11) Å;c=16.0672(8) Å;α=114.301°(6);β=90.322°(4);γ=114.438°(7)。表1中提供了主要氢键的键距和键角。根据收集到的数据绘制ENR-2,6-DHBA·1/2H2O的晶体结构,结果显示,ENR-2,6-DHBA·1/2H2O的不对称单元中包含2个ENR阳离子、2个2,6-DHBA阴离子和1个水分子(图1A);ENR分子内形成O5-H5…O6 (2.580 Å)氢键,2,6-DHBA羧酸基团上的质子转移到了ENR哌嗪环的N原子上,通过N6+-H6…O11-(2.685 Å)和N3+-H3…O7-(2.693 Å)相互作用,其中1个ENR与H2O形成O15-H15A…O1 (2.763 Å)氢键,相邻的2组不对称单元通过2个水分子之间的O15-H15…O15 (2.921 Å)氢键连接成十聚体结构(图1B);相邻十聚体以短接触作用形成1个无限的1D链,1D链通过短接触作用形成2D层(图1C);2D层通过短接触作用、范德华力等形成3D结构(图1D)。

表1 ENR-2,6-DHBA·1/2H2O的氢键参数Table 1 Hydrogen bonding distance and angles for ENR-2,6-DHBA·1/2H2O

续表

A,不对称单元;B,十聚体;C,二维结构;D,三维结构 A,Asymmetric unit;B,Decamer;C,2D structure;D,3D structure图1 ENR-2,6-DHBA·1/2H2O晶体结构Fig.1 Crystal structure of ENR-2,6-DHBA·1/2H2O
2.2 PXRD分析
ENR的PXRD图谱中在衍射角度为6.9°、8.4°、9.5°、14.3°、14.7°、17.1°处具有特征峰,2,6-DHBA在7.7°、11.9°、14.4°、25.1°、27.2°有特征峰,而ENR-2,6-DHBA·1/2H2O的图谱上原料药的特征峰消失,并在6.1°、8.7°、10.7°、12.9°、17.5°、19.5°、26.0°、26.8°、27.9°处出现新的特征峰(图2),形成物的衍射峰与初始物质均不同,证实了新物质的形成[13]。此外,试验制得的ENR-2,6-DHBA·1/2H2O的PXRD图谱与SXRD数据模拟的图谱一致,进一步证实了新物质与培养的单晶相同。
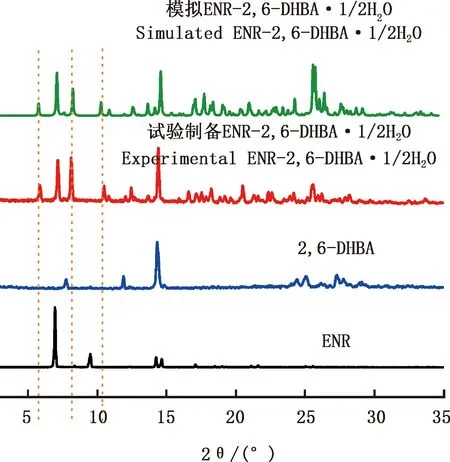
图2 ENR、2,6-DHBA、ENR-2,6-DHBA·1/2H2O及模拟的ENR-2,6-DHBA·1/2H2O的PXRD图谱Fig.2 PXRD diagram of ENR,2,6-DHBA,ENR-2,6-DHBA·1/2H2O and simulated ENR-2,6-DHBA·1/2H2O
2.3 FTIR分析
ENR、2,6-DHBA和ENR-2,6-DHBA·1/2H2O的FTIR结果见图3。由图3可知,ENR在1 736和1 628 cm-1处的吸收峰是羧基和吡啶酮环中的C=O的伸缩振动峰;2,6-DHBA在1 680 cm-1处的吸收峰是羧基中C=O的伸缩振动峰,在3 470 cm-1处的吸收峰是羧基中-OH的伸缩振动峰。形成盐后,2,6-DHBA中1 680 cm-1处C=O的吸收峰向高波数移动到1 725 cm-1,表明2,6-DHBA羧基的质子转移到ENR哌嗪环的N原子上。ENR-2,6-DHBA·1/2H2O红外光谱中3 450 cm-1处出现的宽频带是水分子中-OH的伸缩振动峰[14]。
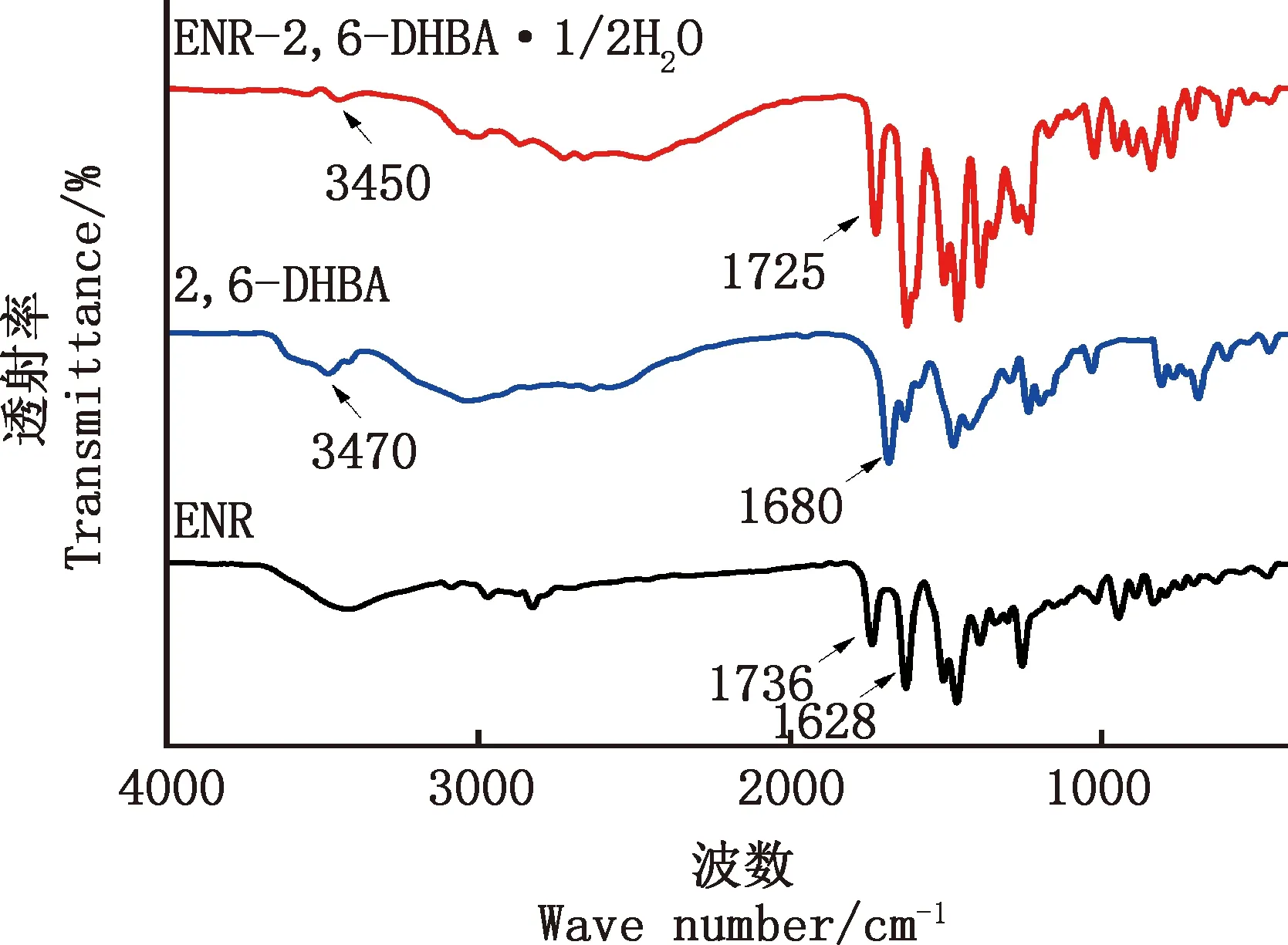
图3 ENR、2,6-DHBA和ENR-2,6-DHBA·1/2H2O的FTIR图谱Fig.3 FTIR spectra of ENR,2,6-DHBA and ENR-2,6-DHBA·1/2H2O
2.4 热分析
ENR、2,6-DHBA和ENR-2,6-DHBA·1/2H2O的DSC、TGA结果见图4。由图4可知,ENR在224 ℃时出现明显的吸热峰,这是ENR的熔点峰,在290 ℃时出现热失重,这与TGA结果相吻合;ENR-2,6-DHBA·1/2H2O在51 ℃开始出现1个小的吸热峰,且TGA图中在51 ℃出现第1个热失重过程,约失重1.68%,经计算,ENR-2,6-DHBA·1/2H2O分子中的1/2H2O占总分子质量的1.72%,证实此失重对应晶体结构中存在的半分子水;DSC中在253 ℃开始出现吸热峰,对应其熔点,TGA图中在251 ℃出现第2个失重过程,失重约30%,经计算ENR-2,6-DHBA·1/2H2O分子中2,6-DHBA占总分子质量的30%,因此该失重对应2,6-DHBA的分解;295 ℃出现第3个失重为ENR的失重过程。

图4 ENR、2,6-DHBA和ENR-2,6-DHBA·1/2H2O的DSC(A)及TGA(B)曲线Fig.4 DSC (A) and TGA (B) curves of ENR,2,6-DHBA and ENR-2,6-DHBA·1/2H2O
2.5 SEM观察
ENR形貌呈现为棱角分明的长条块状(图5A),2,6-DHBA呈现出无规则的碎块状或碎条状(图5B),而ENR-2,6-DHBA·1/2H2O是表面光滑的棒状(图5C)。可见制备的新盐形貌不同于ENR和2,6-DHBA。
2.6 标准曲线的绘制
使用高效液相色谱对ENR待测液进行测定,在5.45 min有样品峰(图6),测得峰面积值后,以浓度为横坐标、峰面积为纵坐标绘制标准曲线(图7),回归方程为:y=―30 678.32+165 472.57x,R2=0.9998,在0.3125~80 μg/mL浓度范围内线性良好。

图6 ENR标准品液相色谱图Fig.6 Liquid chromatogram of ENR standard

图7 HPLC测定ENR的标准曲线Fig.7 Standard curve of ENR of HPLC determination
2.7 溶解速率及溶解度测定
ENR和ENR-2,6-DHBA·1/2H2O在37 ℃、pH 6.8磷酸缓冲液中的粉末溶出结果显示,ENR原料药在20 min时浓度达到0.67 mg/mL,随后浓度开始下降,6 h后降至0.35 mg/mL,达到平衡(图8)。对溶出试验后的固体粉末进行PXRD检测发现,ENR在溶出后发生转变,在水溶液中会转变成溶解度更低的ENR·6H2O[15];ENR-2,6-DHBA·1/2H2O盐在溶出1 h后表观溶解度为0.65 mg/mL,直至8 h浓度依然保持稳定,此时溶解度是ENR的1.86倍,对溶出后的残渣进行PXRD检测发现,ENR-2,6-DHBA·1/2H2O在溶出介质中没有发生转变(图9)。说明ENR-2,6-DHBA·1/2H2O在水溶液中具有较好的稳定性。

图8 ENR和ENR-2,6-DHBA·1/2H2O的粉末溶出曲线Fig.8 Powder dissolution curves of ENR and ENR-2,6-DHBA·1/2H2O
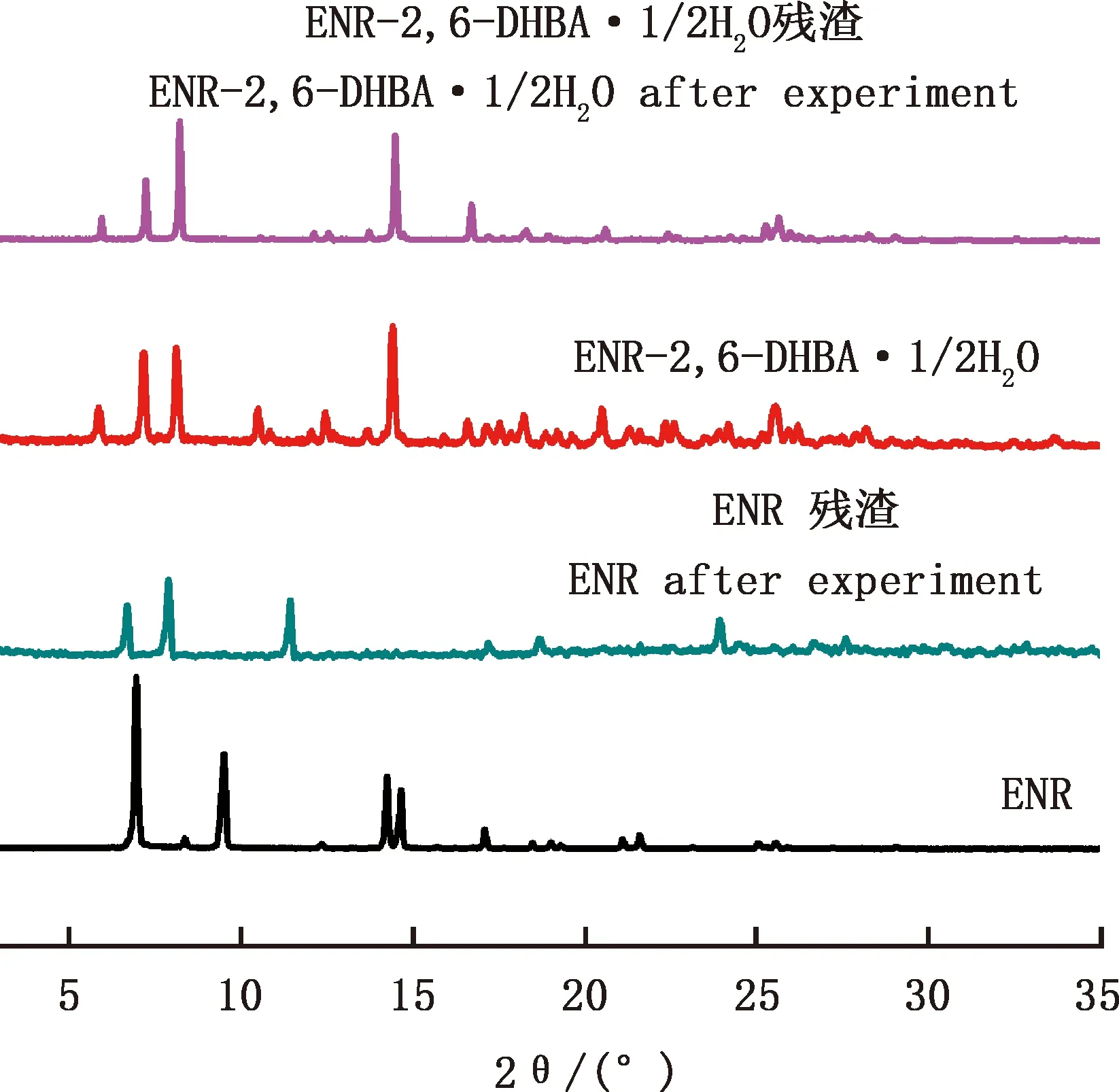
图9 ENR和ENR-2,6-DHBA·1/2H2O试验后残渣的PXRD图谱Fig.9 PXRD diagram of ENR and ENR-2,6-DHBA·1/2H2O after experiment
3 讨 论
ENR是化学合成的抗菌药物,杀菌作用强,进入机体后易被组织吸收,在体内分布广泛[16]。但ENR在中性水溶液中溶解度较低,低水溶性极大地限制其新型制剂的开发,另外,ENR属于浓度依赖性抗菌药物[17],在临床使用时为了达到治疗所需血药浓度,通常会投入较大的使用剂量,会增大毒副作用。药物成盐是指在适宜的溶剂中药物活性成分与带有相反电荷的组分均电离,然后两者以离子键相结合,以盐的形式结晶析出的过程[18-19]。将药物活性成分和一种共形成物制备成盐可以解决药物溶解度低的问题[20]。
3.1 ENR新盐的晶体结构与表征
本研究以ENR为药物活性成分,2,6-二羟基苯甲酸为共形成物,采用混悬法制备了一种ENR新盐。PXRD图谱显示了旧峰的消失和新峰的出现,证实新物质的生成。单晶结构解析可对晶体的晶型进行鉴别,确定晶体结构中原子、分子、离子在三维空间的排布规律[21],经单晶结构解析可知该新物质ENR-2,6-DHBA·1/2H2O,属于三斜晶系P-1空间群,结构中ENR、2,6-DHBA和H2O的比例为2∶2∶1,由于存在质子的转移,因此制备的新物质是一种盐。
FTIR具有灵敏度高、光谱范围广和分辨率高等优点[22],已广泛用于共晶、盐、溶剂化物/水合物的鉴定以及区分单组分和多组分晶体的不同多晶型[23]。在FTIR中,当有分子间相互作用时,相关官能团的伸缩振动峰会发生偏移。Wang等[24]制备了阿司匹林-川芎嗪药物-药物共晶,FTIR结果显示该共晶中阿司匹林在3 487.04 cm-1处羧基中-OH的伸缩振动峰以及川芎嗪在1 411.34 cm-1处对应哌嗪C=N的伸缩振动峰均消失了,这与阿司匹林和川芎嗪之间形成分子间氢键(O-H…N)有关。本研究制备的新盐由于质子的转移(形成N+-H…O-)导致2,6-DHBA羧基中C=O在1 680 cm-1处的吸收峰偏移到了1 725 cm-1处。
热性质是评估固态药物物理化学转变过程(如形成过程、多态转变和分解)的参数[25],方法通常包括TGA和DSC。通过控制温度的升降,测定药物粉末随温度变化而产生的理化性质方面的改变,如脱水、升华、熔化、氧化、分解及晶型转变,也可以作为检测盐/共晶纯度的一个辅助手段。周凯等[26]制备了黄连素-染料木素有机盐水合物,晶体结构中包含水分子和乙醇分子,在TGA分析中,有机盐水合物在30~150 ℃失去10%的质量,这与水和乙醇分子总质量分数(10.1%)相吻合,且在DSC图中,该水合物在30~150 ℃出现了1个较宽的吸热峰,对应着失去水和乙醇的过程。本研究中,ENR-2,6-DHBA·1/2H2O的TGA图中在100 ℃之前出现了1个小的失重,约为1.68%,对应晶体结构中的半个水分子(经计算约为总质量分数的1.72%),且在DSC图中也出现了1个宽而小的吸热峰,对应了水分子的蒸发。
3.2 ENR新盐的溶出速率及溶解度
固态药物的表观溶解度和溶出度对药物开发和质量控制至关重要[27]。药物盐/共晶是提高难溶性药物溶解度、溶出速率的有效方法,其机制有两种,首先,晶体晶格能的降低可引起溶解度的增加;其次,具有更好亲水性的共形成物更有可能增加所得盐/共晶的溶剂亲和力[28-29]。坎地沙坦是一种水溶性差的药物[30],Srivastava等[31]将坎地沙坦和对羟基苯甲酸甲酯制备成共晶,采用摇瓶法测定溶解度,坎地沙坦的溶解度为2.03 mg/mL,而共晶的溶解度为14.12 mg/mL,表明坎地沙坦形成共晶后溶解度提高了约6倍。在溶解度研究后对得到的固体粉末干燥并进行PXRD测定,固体粉末的衍射峰与试验前没有发生变化,表明它在48 h后仍是稳定的。ENR在溶出20 min时浓度达到0.67 mg/mL,但随后的时间点浓度逐渐下降,降至0.35 mg/mL达到平衡,根据溶出曲线的变化推测ENR在溶出过程中可能发生了转变,收集8 h溶出后的固体粉末测定PXRD发现衍射峰发生了变化,溶出后的物质与ENR·6H2O的PXRD衍射峰吻合,判断ENR在溶出介质中接触水之后转变成了溶解度更低的ENR·6H2O。而ENR-2,6-DHBA·1/2H2O在溶出1 h后达到平衡没有出现浓度下降的情况,此时的表观溶解度为0.65 mg/mL,是ENR的1.86倍,通过对溶出后的固体粉末测定PXRD发现,ENR-2,6-DHBA·1/2H2O溶出前后没有发生变化,证明该盐在水中是稳定的。
4 结 论
试验利用晶体工程技术,采用混悬法制备了ENR-2,6-DHBA·1/2H2O新盐,通过溶剂挥发法培养了单晶,晶体结构显示ENR-2,6-DHBA·1/2H2O属于三斜晶系,P-1空间群,ENR与2,6-DHBA通过N+-H…O-相互作用。新盐的外貌形态呈光滑的棒状,熔点为253 ℃。平衡时的表观溶解度是ENR的1.86倍,说明ENR-2,6-DHBA·1/2H2O可提高ENR的溶解性。
猜你喜欢
铝加工(2022年1期)2022-11-24
医药导报(2022年5期)2022-04-27
兵器装备工程学报(2022年3期)2022-04-08
人工晶体学报(2021年10期)2021-11-26
粉末冶金技术(2021年3期)2021-07-28
兵器装备工程学报(2021年5期)2021-06-02
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21
中国科技纵横(2018年3期)2018-03-15
中学生数理化·高三版(2016年2期)2016-09-10
