
原位拉曼光谱研究电催化反应过程
2022-10-19 07:42阿尧林王耀辉董金超李剑锋
广西师范大学学报(自然科学版) 2022年5期
阿尧林, 王耀辉, 董金超*, 李剑锋,
(1.厦门大学 能源学院, 福建 厦门 361102; 2.厦门大学 化学化工学院, 福建 厦门 361005)
随着经济社会的快速发展,能源危机加剧,人们对新型能源的需求与日俱增。因此,开发高效、清洁、可再生的能源显得尤为重要。其中,电化学技术作为一种重要的能量转换途径,可有效实现化学能与电能的相互转换。例如,燃料电池技术通过阴极的氧还原反应(ORR)和阳极的氢氧化反应(HOR)可以高效地将氧气和氢气中的化学能转化为电能[1-2]。而通过电解水析氢反应(HER)和析氧反应(OER),人们则可以利用电能将水分解为氢气和氧气[1-3]。除此之外,利用电化学还原技术还可以将二氧化碳气体转换为价值更高的碳氢化合物,这也是实现碳中和的重要途径之一[1,4]。
固/液界面的电催化反应过程往往涉及多种表面吸附物种和众多寿命短、浓度低、信号弱的反应中间物种,使得其界面反应机理非常复杂[2, 5-7]。由于缺乏直接和实时的原位实验证据,固/液界面电催化反应机理和构效关系仍不明确,限制了高效催化剂的理性设计和开发。传统的电化学表征技术主要是从宏观层面上提供固/液界面电化学反应的平均信息,很难直接给出关键吸附/反应物种在电催化反应过程中的确切存在状态及演变信息。因此,急需开发先进的原位表征技术,在分子乃至原子层面深入研究电催化反应过程及相关中间物种的信息。作为高灵敏的原位表征技术,增强红外光谱和X射线吸收光谱等可提供表界面吸附分子的结构信息或电子结构信息等,可以用于进一步揭示反应机理,为设计高效催化剂提供实验和理论基础[8-9]。但受技术或反应体系等限制,例如X射线吸收光谱成本高昂,而红外光谱受水的干扰大、低波数区域(<1 000 cm-1)检测不足等,使得上述技术在电催化反应机理的研究中受到一定制约。
作为一种分子指纹光谱技术[10],表面增强拉曼光谱(SERS)的出现[11-12],极大推动了拉曼光谱在电催化反应研究中的应用。SERS技术具有极高的表面检测灵敏度且不受溶液水的干扰,可有效检测电极表面的吸附分子/物种信息,非常适宜于研究表界面电催化反应过程。但经过长期的研究,人们发现只有在粗糙的金、银、铜和少数特定的金属表面才具有强的SERS增强效应[13-14],这在一定程度上也限制了SERS技术在表面科学中的进一步应用和发展。而壳层隔绝纳米粒子增强拉曼光谱(SHINERS)[15]的发明突破了传统SERS在材料和表面形貌普适性差的长期局限,使得人们可以在任意材料和任意表面获得增强拉曼信号,有效拓宽了增强拉曼光谱技术的应用领域,并成功应用于模型单晶体系和实际纳米催化剂的原位研究中。接下来,本文将分别介绍不同的原位增强拉曼光谱技术及其在重要电催化反应中的应用,最后进行简单的总结展望。
1 电催化反应的原位拉曼光谱研究策略
电磁场增强理论认为,在合适激发光的照射下,具有一定纳米级粗糙度的金属基底或金属纳米粒子表面产生的极强电磁场,可极大增强表界面吸附物种的拉曼信号,该现象也被称为SERS效应。相较于常规拉曼光谱技术,SERS技术具有极高的表面检测灵敏度[16],可以实现单分子信号的检测分析,这也使得利用SERS有效检测电催化反应(例如ORR、OER等)表界面痕量反应/中间物种的直接拉曼信号成为可能。但传统SERS技术无法直接获得燃料电池常用非等离激元的Pt、Pd、Ru等过渡金属材料表面的拉曼信号,人们发展出一种“借力”策略(图1b),即通过在Au纳米粒子(Au NPs)表面上沉积/包覆过渡金属壳层,利用内核Au NPs产生极强的电磁场,增强其外层过渡金属表面吸附物种的拉曼信号,从而实现对于不同过渡金属材料表界面电催化反应过程的原位监测[17]。

图1 SERS工作原理Fig.1 SERS working principle diagram
设计具有高活性和高选择性的催化剂需要深入理解催化过程中的反应机制。原子级平整的单晶电极作为表面科学研究中最重要的模型体系,具有确定的表面结构和确定的能级状态[18],被广泛应用于研究电催化反应机理。但传统的SERS技术和“借力”策略均无法有效获得单晶表面的增强拉曼信号。原位电化学SHINERS作为一种具有高表面检测灵敏度的光谱分析技术,在原位研究单晶表面电催化反应机理方面表现出巨大优势。在SHINERS技术中,通过在Au NPs表面包覆一层极薄致密的惰性SiO2或者Al2O3壳层,可形成壳层隔绝纳米粒子(SHINs)。SHINs是一种特殊的核壳纳米结构,内核Au NPs具有强的SERS增强能力,而外层惰性、致密和超薄的SiO2壳层,可有效隔绝内核Au NPs不干扰外界催化反应过程,从而获得更真实的电催化反应信息。SHINERS技术的增强效果可通过改变内核纳米粒子材质(Au、Ag和Cu)、粒径(50~200 nm)以及减少壳层厚度来提高,一般壳层厚度需小于2 nm且无针孔。SHINERS具有通用性强、表面灵敏度高、可获得低波数信息、热稳定性好等优点,是一种理想的电催化原位表征技术。在电催化研究过程中,通过将SHINs组装在单晶电极表面,如图1c所示,利用SHINs与金属电极之间的耦合作用产生增强电磁场来放大拉曼光谱信号,从而获得附近痕量反应/中间物种的拉曼信息[19-20]。
由于实际过渡金属纳米催化剂粒径非常小,几乎无拉曼增强效应,我们通过发展实际催化剂的可控组装技术,将原位增强拉曼光谱的研究对象从模型单晶表面进一步拓展到实际纳米催化体系。具体来说,通过表面修饰或静电组装策略,将实际催化剂组装在SHINs表面,从而构建SHINERS-卫星结构[20]。如图1d所示。在SHINERS-卫星结构中,SHINs中的Au NPs能有效发挥信号放大器的作用,增强催化剂表面的拉曼信号;而惰性的SiO2壳层则能有效隔绝Au NPs干扰,保证测得的拉曼信号来自于催化剂表面反应过程,从而实现实际催化剂表面电催化反应过程的原位表征。
2 电催化反应过程的原位拉曼光谱研究
2.1 氧还原反应(ORR)
氧还原反应(ORR)作为质子交换膜燃料电池最重要的阴极反应,其高的过电位和缓慢的动力学过程,严重制约着燃料电池技术的进一步发展和应用[21]。ORR反应是一个涉及到二电子和四电子转移的反应过程。而在二电子过程中,反应会生成过氧化氢(H2O2),导致燃料电池效率变低,并且影响隔膜的稳定性[22]。所以,在燃料电池中,阴极反应的理想途径是四电子过程,四电子过程又分为缔合机理(associative mechanism)和解离机理(dissociative mechanism)[23]。以酸性条件下的ORR反应为例,缔合机理的具体步骤为[2]:
(1)
(2)
OOH*+ H++ e-→O*+H2O,
(3)
O*+ H++ e-→OH*,
(4)
OH*+ H++ e-→H2O。
(5)
其中*代表催化剂表面的活性位点。而解离机理的具体步骤为:
O2+ 2*→2O*,
(6)
2O*+ 2H++ 2e-→2OH*,
(7)
2OH*+ 2H++ 2e-→2H2O。
(8)
然而,ORR反应过程不仅牵涉到众多常规技术极难捕获的表面吸附物种和反应物种,并且存在多种可能的反应途径,使得其机理解释极为复杂。所以,研究ORR反应机理关键在于利用高灵敏的原位谱学技术捕获确定ORR反应过程重要中间物种的直接光谱证据。
2.1.1 Pt单晶电极
目前,Pt基材料仍是最优异的ORR催化材料。相对于多晶Pt电极,单晶Pt电极表面原子结构均一,易于和理论模拟相结合[24],可以从更深层次揭示ORR反应机理,为催化剂的设计提供更好的指导。本课题组[25]利用原位SHINERS技术,系统研究了低指数Pt单晶电极表面的ORR过程(图2)。由电化学活性表征可知,在氧气饱和的0.1 mol/L HClO4溶液中,3个低指数铂单晶电极的ORR活性为:Pt(111) >Pt(110) >Pt(100)。其中图3(a)为酸性条件下,Pt(111)单晶表面ORR反应过程的原位电化学拉曼光谱。随着ORR反应的进行,首先在约732 cm-1附近观测到和过氧物种相关的直接拉曼光谱证据,而在相应的氚代同位素实验中,该峰偏向低波数区域。结合理论计算,进一步确认该峰归属为ORR反应关键中间物种OOH*的特征拉曼谱峰。图3(b)为酸性条件下Pt(110)晶面上ORR反应的原位电化学拉曼光谱结果,结合重水(D2O)和18O2的同位素验证以及理论计算,确认位于约1 080 cm-1的拉曼峰属于ORR关键中间物种OH*的特征拉曼谱峰。

图2 Pt单晶表面EC-SHINERS的模型图Fig.2 Model of EC-SHINERS on Pt single crystal surface

图3 Pt单晶表面ORR过程的原位电化学SHINERS光谱Fig.3 In-situ electrochemical SHINERS spectra of ORR process on Pt single crystal surface
高指数Pt单晶因其优异的催化活性也受到人们的关注,为了进一步探究其ORR机理,本课题组[26]利用SHINERS技术,原位研究了高指数铂单晶表面的ORR过程。图3(c)显示了Pt(311)在0.1 mol/L HClO4中的电化学拉曼光谱,通过结合同位素的验证和理论计算,首次在高指数铂单晶表面发现ORR关键中间物种OH*(1 041 cm-1)和OOH*(765 cm-1)的直接光谱证据。结合理论计算结果,发现由于Pt(311)晶面对OOH*物种具有过强的吸附致其更难从电极表面解离脱附,导致Pt(311)晶面的ORR活性较低。综上表明,不同的Pt(hkl)单晶电极由于具有不同的表面结构,导致其对中间物种的吸附能力不同,进而影响其ORR催化活性,反映出中间物种对单晶界面ORR反应过程的构效关系有重要的影响。基于铂单晶表面ORR原位拉曼光谱研究,确认了在Pt单晶表面的ORR过程为吸附的O2通过电子质子转移形成OOH*物种,然后OOH*解离生成OH*与O*,OH*物种再进一步与H结合生成H2O的四电子反应过程(图4)。
2.1.2 铂(Pt)合金催化剂及多晶电极
目前,将Pt与过渡金属形成合金化结构是制备高活性、高稳定性ORR催化剂最有效的策略之一[27],不仅能有效提高Pt的利用率,还能通过配体或者应变效应,进一步提高其ORR活性。然而,由于缺乏直接的光谱证据,人们对合金催化剂界面的ORR机理还存在一定争议。本课题组[28]采用“借力”策略,通过在Au内核外面沉积均匀的PtNi壳层,并改变壳层中Ni的含量,研究其对ORR过程中含氧物种吸附构型变化的影响。其中内核Au NPs产生的电磁场可有效放大PtNi壳层表面ORR过程中间物种的拉曼信号,从而获得高质量的拉曼谱图。通过同位素验证和理论计算,确认了PtNi催化剂表面的ORR过程中,有OOH*中间物种(732 cm-1)的存在(图5(a))。通过改变PtNi壳层中Ni的含量(图5(b)),发现OOH*物种的拉曼峰位置随着Ni含量的增加而红移。结合理论模拟,确认Ni含量会影响OOH*在催化剂表面的结合能,Ni元素的加入更利于ORR过程中的电子转移,提高ORR活性。
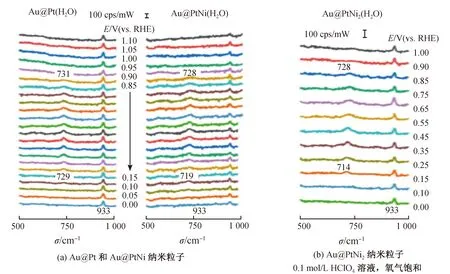
图5 纳米粒子的原位拉曼光谱Fig.5 In situ Raman spectra of NPs

图6 氧还原过程中Pt3Co纳米催化剂的原位电化学SHINERS光谱Fig.6 In situ EC-SHINERS spectra of ORR on Pt3Co NPs

2.2 氢氧化反应(HOR)
氢氧化反应作为质子交换膜燃料电池的阳极反应,其在酸性介质和碱性介质中有不同的微观反应路径。在酸性介质中可能遵循Tafel/Volmer或者Heyrovsky/Volmer步骤[31]。
Tafel步骤:
H2→2Had。
(9)
Heyrovsky步骤:
H2→Had+ e-+ H+。
(10)
Volmer步骤:
Had→H++ e-。
(11)
两条路径都是H2吸附到催化剂表面,成为吸附的氢(Had),然后释放一个电子变为氢离子(H+),因此,在HOR反应中,也将氢的结合能作为评判反应活性的描述符。
而在碱性介质中的路径略有不同[32]。
Tafel步骤:
H2→ 2Had。
(12)
Heyrovsky步骤:
H2+ OH-→ Had+ H2O + e-。
(13)
Volmer步骤:
Had+ OH-→ H2O + e-。
(14)
在HOR过程中,由于吸附的羟基在催化剂表面极低的覆盖率,导致普通方法很难监测到该物种。为探究HOR的反应路径,本课题组[33]采用“借力”策略,制备Au@PtNi核壳纳米结构(图7),由电镜表征能看到,PtNi催化材料均匀沉积在Au内核上,内核金纳米粒子产生的极强电磁场能有效增强PtNi材料表面的拉曼信号。相应的电化学极化曲线显示PtNi材料有着更高的HOR活性(图8),表明Ni的掺杂有利于HOR活性的提升。之后,基于此结构利用电化学拉曼光谱原位研究PtNi材料上的HOR反应过程。图9表明,在位于约778 cm-1处观察到明显的拉曼谱峰,并通过同位素验证以及理论计算,认为该峰属于Ni-Ni桥位吸附的OH物种(OHad)振动引起的。通过理论计算得出,OH-在溶液中的吉布斯自由能要高于吸附的OH(OHad)(图10)。因此,电极表面吸附的OH(OHad)更有利于HOR反应过程的进行。结合实验结果与理论计算(图11),可以认为在纯Pt表面发生的HOR过程,仅有Had与溶液中OH-反应生成H2O;而在PtNi表面,Ni的掺杂有利于OH的吸附生成OHad,可与Had反应直接生成H2O。综上所述,Ni表面吸附的OHad物种和Pt表面吸附的Had物种反应生成水,促进了HOR活性的提升。

图7 Au@PtNi纳米粒子的“借力”策略示意以及电镜图谱Fig.7 Au@PtNi NPs schematic diagram and electron microscopy of “borrowing” strategy
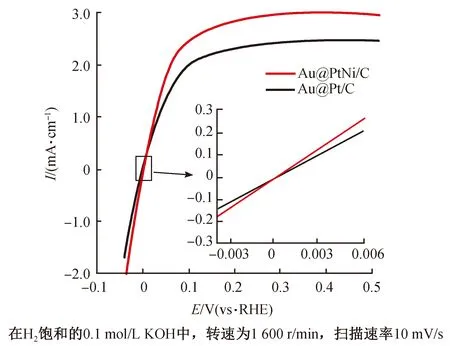
图8 Au@PtNi/C和Au@Pt/C的HOR极化曲线Fig.8 HOR polarization curves of Au@PtNi/C and Au@Pt/C

图9 Au@PtNi在氢氧化过程中表面的原位SERS光谱Fig.9 In situ SERS spectra of the HOR on and Au@PtNi surfaces
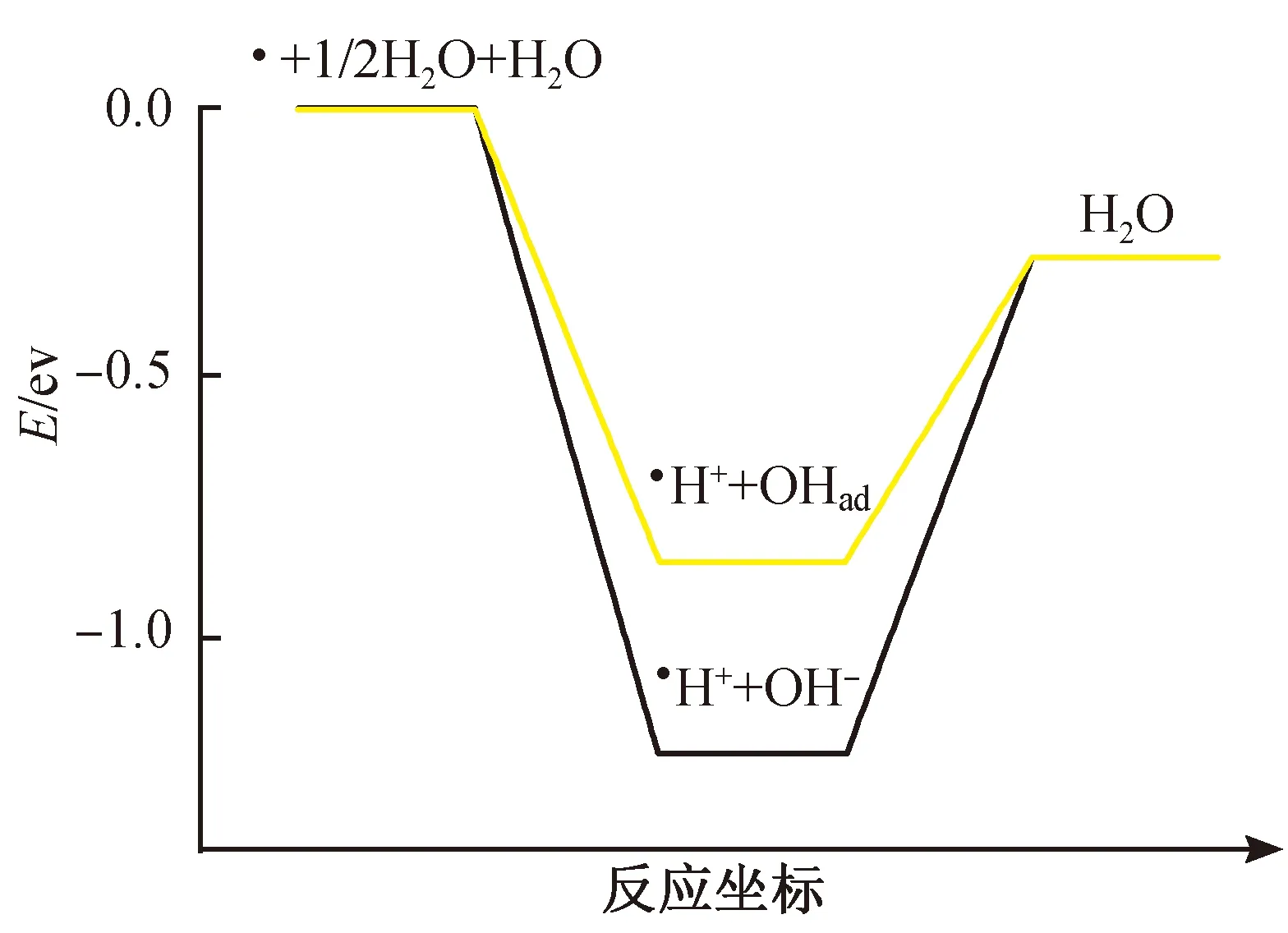
图10 HOR过程的吉布斯自由能分布Fig.10 Gibbs free energy distribution diagram of HOR process
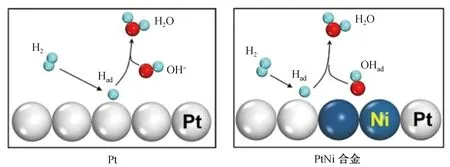
图11 碱性条件下表面模拟的HOR路径Fig.11 Simulated HOR paths under alkaline conditions
2.3 氧析出反应(OER)
电催化裂解水产生氢气和氧气,是解决能源危机的可行技术之一。在阳极发生的氧析出反应(OER),存在反应速率缓慢、过电位大等问题,需要高效催化剂来加速反应的发生。在早期阶段,贵金属材料因其优异的催化活性和稳定性,得到了学者的广泛关注,其中钌(Ru)和铱(Ir)的氧化物催化剂在酸性和碱性条件下都展现出优异的OER性能,但其较高的成本阻碍了大规模的应用[34]。因此,发展非贵金属OER催化剂显得尤为重要。目前过渡金属及其氧化物,以及多元金属催化剂的制备等已成为制备高效OER催化剂的有效方法之一[35]。为了理性设计性能更优异的催化剂,需要对OER反应过程和活性位点有更深入的研究和理解。
一般来说,OER过程在酸性溶液和碱性溶液中有不同的反应机制[36]。具体如下。
酸性条件下:
M + H2O→MOH + H++ e-,
(15)
MOH + OH-→MO + H2O + e-,
(16)
2MO→2M + O2,
(17)
MO + H2O→MOOH + H++ e-,
(18)
MOOH + H2O→M + O2+ H++ e-。
(19)
碱性条件下:
M + OH-→MOH,
(20)
MOH + OH-→MO + H2O,
(21)
2MO→2M + O2,
(22)
MO + OH-→MOOH + e-,
(23)
MOOH + OH-→M + O2+ H2O。
(24)
上述反应式中M为金属,通过碱性和酸性反应能得到,形成O2的方式有2种:一种途径是MO物种直接分解形成O2;另一种途径是先形成MOOH物种,再进一步分解形成O2。但人们对具体的OER反应机理仍存在很多争议。
为了有效揭示OER反应机理,人们也利用增强拉曼光谱技术研究不同材料界面的OER反应过程。有研究表明[37],通过将Ni-Fe催化剂沉积在Au电极表面,然后利用增强拉曼光谱技术研究Ni-Fe催化剂界面的OER过程,在Ni-Fe催化剂表面观测到含有类似NiOOH物种的直接光谱证据(图12),并且由于Fe元素的存在,改变了Ni-O物种的局部环境,进而提高OER催化活性。另一项研究则通过原位拉曼光谱研究了Ni基催化剂界面的OER过程[38],研究者发现在电位逐渐升高的过程中,Ni基纳米粒子(Ni NPs)经不可逆的过程先转变为ɑ-Ni(OH)2/NiO,再经可逆过程转变为γ-NiOOH,其中γ-NiOOH为OER过程中的主要活性位点(图13)。在催化剂表面发生的重构过程改变了Ni基材料的电子结构,从而促进电子从活性位点向本体转移,这也是OER活性提升的关键因素。此外,有学者研究在碱性条件下,通过将Co3O4沉积在Au核表面,利用原位拉曼光谱表征其OER反应过程[39]。如图14结果所示,随着电位升高,Co3O4氧化物逐渐被氧化成CoO(OH),且由于Au内核表面沉积的Co3O4存在,增加了Co4+位点在Au核表面的沉积,使该催化剂的OER活性提高。

图12 Ni薄膜沉积在粗糙Au基底上的原位拉曼光谱Fig.12 In situ Raman spectra for Ni films atop a roughened Au substrate
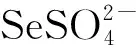
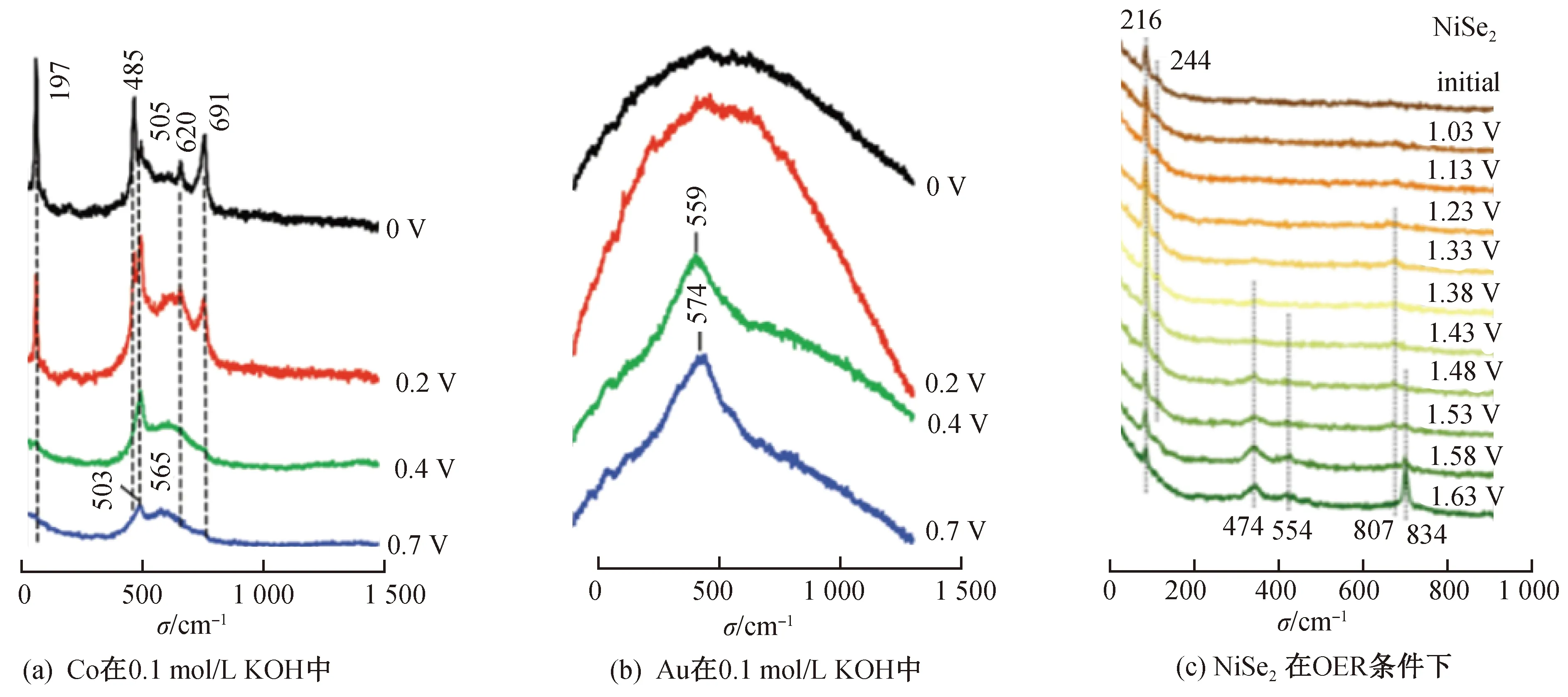
图14 Co、Au和NiSe2的原位拉曼光谱Fig.14 In situ Raman spectra of Co, Au and NiSe2
2.4 氢析出反应(HER)
氢析出反应(HER)作为电催化裂解水的阴极反应,在不同的电解质中有不同的反应路径。在酸性介质中,因为存在源源不断的质子,可促进HER过程;但是在碱性溶液中,需要额外的能量分解水产生质子,降低了反应的整体速率,同时也存在较大的过电位。目前认为,碱性条件下的HER过程一般可分为如下所示的Volmer-Heyrovsky步骤和Volmer-Tafel步骤[42]。
Volmer步骤:
H2O + e-→H*+ OH-。
(25)
Heyrosky步骤:
H2O + e-+ H*→H2+ OH-。
(26)
Tafel步骤:
H*+ H*→H2。
(27)
但对碱性条件下具体的HER反应机理人们还存在一定争议。为了研究碱性条件下的HER反应机理,研究者也基于原位拉曼光谱探究了不同催化剂表面的HER反应过程。

图15 在OCP和恒电位下的原位拉曼光谱Fig.15 In situ Raman spectra at OCP and constant potentials
Zhao等[43]首先制备单原子碘化镍催化剂,并将其应用于碱性溶液中的电催化HER过程,之后利用原位电化学拉曼光谱研究该催化剂表面的HER反应过程。基于原位拉曼光谱结果,在HER过程中确认催化剂表面吸附的氢(Hads)与碘原子(I)通过Volmer步骤结合形成I-Hads中间体,而裂解产生的OH-与镍(Ni)结合,促进了HER过程(图15)。除此之外,过渡金属硫族化合物展现出优异的HER活性[44],但还缺乏在原子层面揭示其HER的过程,利用光谱捕获催化过程中产生的中间体信息是揭示其催化机理的关键。根据研究结果[45],将湿化学合成法制备单层MoS2包覆在Ag多面体表面,在酸性条件下,通过原位拉曼光谱监测其HER过程。通过同位素验证的拉曼光谱表明,在位于约2 532 cm-1处的拉曼峰为S—H的伸缩振动(图16),其中,MoS2中的S原子为HER过程中的活性位点。

图16 HER过程下Ag@MoS2的电化学SERS光谱Fig.16 EC-SERS spectra of HER on Ag@MoS2
2.5 二氧化碳还原反应(CO2RR)
电催化二氧化碳还原是减少二氧化碳、实现碳循环的有效方法之一,CO2RR可有效将CO2转化成价值更高的碳氢化合物,这也是解决能源危机的关键[4,46]。然而,CO2RR反应过程和物种极其复杂,不同表面吸附物种/反应物种又与催化剂本身的活性息息相关。因此,捕获CO2RR中间产物的信息是理解该反应并确定其反应路径的关键。
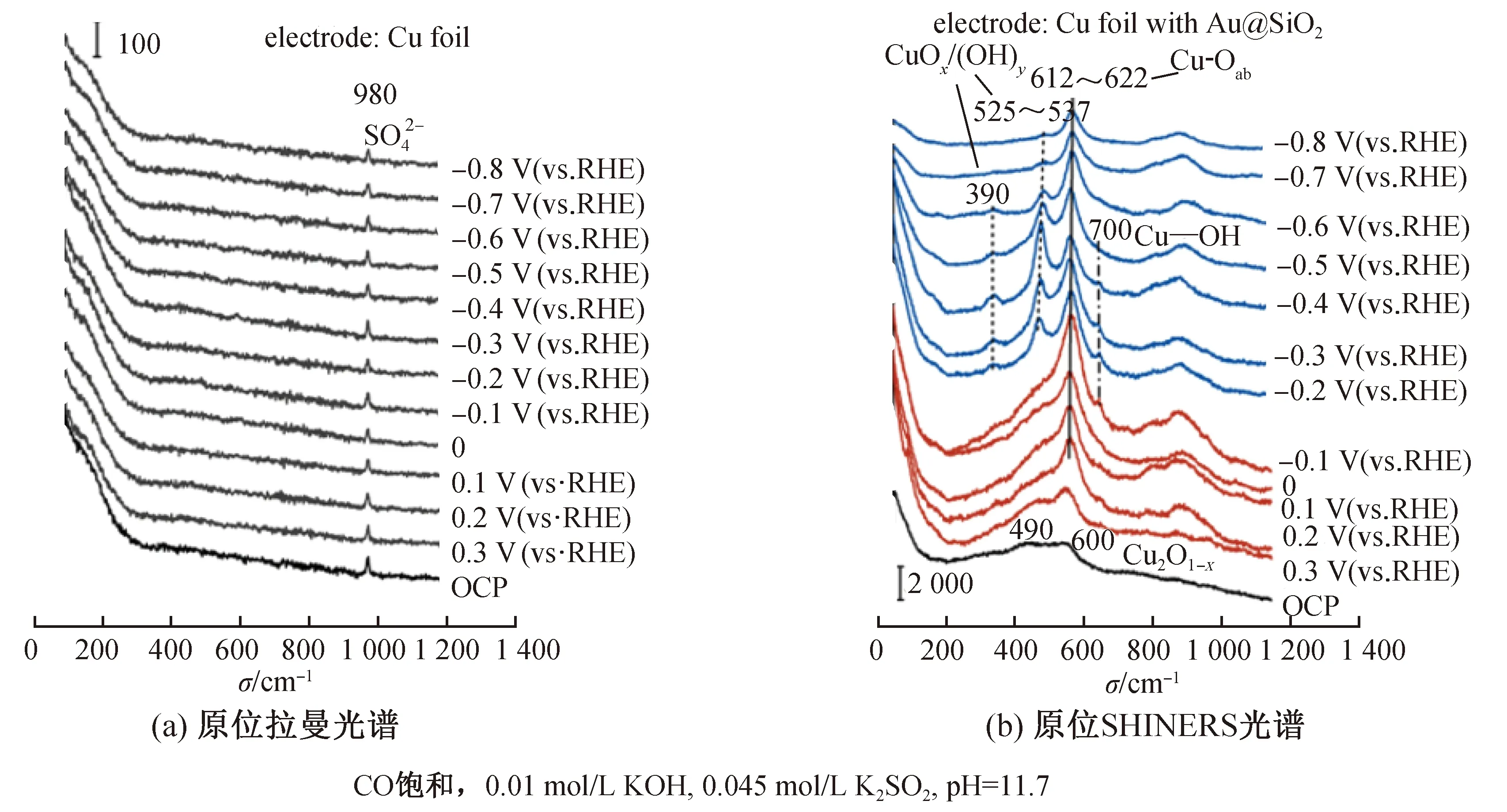
图17 Cu箔的原位拉曼光谱和原位SHINERS光谱Fig.17 In situ Raman spectra and in situ SHINERS spectra of a Cu foil
铜(Cu)是唯一一种具有高选择性、且能够直接将CO2和CO转化为有价值碳氧化合物和碳氢化合物的单质金属。目前,研究者已利用SERS技术对Cu表面CO还原过程进行研究[47],但仅在位于约980 cm-1处观察到溶液本体的峰(图17(a))。之后研究者在Cu箔表面引入SHINERS技术,光谱结果表明,在0.3 V时,位于约612 cm-1处出现Cu-Oad的特征峰,在电位降低至-0.1 V时,该峰强度变强并蓝移至约620 cm-1处,当电位进一步降低至-0.8 V时,该峰强度减弱,并蓝移至约622 cm-1处。除此之外,在位于约515 cm-1处观察到铜氢氧化物与氧化物的物种,以及在位于约700 cm-1处观察到Cu-(OH)x物种的特征拉曼谱峰证据(图17(b))。图18(a)所示为在硅(Si)表面电化学沉积Cu薄膜的CORR电化学拉曼光谱,谱图结果显示,当电位为-0.2 V时,在位于约392 cm-1、约530 cm-1和603 cm-1处出现3条清晰的特征峰,且位于约530 cm-1与约603 cm-1处的峰相对强度与Cu箔表面相反。综上所述,在负电位下,Cu表面均存在多种氧化物和氢氧化物,但Cu箔表面与电化学沉积的Cu薄膜有所差异。
目前,已有大量研究探寻具有较低过电位和较高法拉第效率的CO2RR电催化剂[48]。其中铜基催化剂表现出较好的性能,可采用纳米结构和双金属的策略来改变Cu基催化剂的活性和选择性。Ren等[49]通过制备不同锌(Zn)掺杂的铜(Cu)基双金属催化剂,并结合原位拉曼光谱研究其CO2RR过程的活性以及选择性。当将电位持续控制在-0.85 V时,原位拉曼光谱显示Cu和Zn的氧化峰逐渐减弱,随后,在位于约280 cm-1、约365cm-1和约2 060 cm-1处出现了相应的CO*、Cu—CO的伸缩振动,以及碳氧三键的伸缩振动(图18b),并在金属氧化物峰消失之前并没有出现CO的相关振动信息,表明CO2RR发生在金属表面。
除了Cu基催化剂,金属锡(Sn)上的CO2RR也得到了较多研究,CO2RR法拉第效率也与Sn的形态、化学状态和电解条件息息相关[50]。Vasileff等[51]采用原位拉曼研究SnO2以及Cu-Sn合金催化剂的CO2RR过程,在Sn含量较低时,CO2RR过程中更有利于与C结合的*COOH中间体生成CO,而随着Sn的含量增加,CO的选择性降低,更有利于通过*OCHO的中间体生成甲酸(图19)。

图19 Cu5Sn6和 Cu12Sn的电化学拉曼光谱Fig.19 Electrochemical Raman spectra for Cu5Sn6 and Cu12Sn
3 结语与展望
基于原位拉曼光谱研究电催化反应过程、获得关键反应/中间物种的直接光谱信息,从分子及原子层面揭示电催化反应机理,对高活性、高稳定性的催化剂制备具有重大意义。本文综述原位拉曼光谱在ORR、HOR、OER、HER以及CO2RR中的一些重要应用,并对其研究结果进行较为详细的介绍和分析。虽然经过多年的发展,原位增强拉曼光谱技术有了显著的进步,但不同研究体系/环境下的拉曼技术需要有合适的增强基底,而传统的SHINs增强粒子还存在一定的局限,如在碱性溶液(pH>12)和高温(>300 ℃)条件下不稳定,不能长时间保持其增强活性等。除此之外,在实际的电催化过程中,反应中间体的寿命极短,拉曼光谱采集时间较长,获取其信息可能较为有限。因此,需要发展更加先进的增强拉曼光谱技术用于实际的催化剂领域。
猜你喜欢
辽宁省博物馆馆刊(2021年0期)2021-07-23
中国粮油学报(2018年12期)2018-03-19
光学精密工程(2016年1期)2016-11-07
中国科技信息(2016年19期)2016-10-25
中国科技信息(2016年6期)2016-08-31
中国科技信息(2015年24期)2015-11-07
中国科技信息(2015年23期)2015-11-07
原子能科学技术(2014年3期)2014-02-28
郑州大学学报(理学版)(2013年3期)2013-03-11
物理与工程(2013年2期)2013-03-11
