
不同提取和改性方式小粒咖啡果皮果胶的理化性质及其抗氧化活性
2022-10-17 04:56李晓娇汪玉洁杨丽华唐金山宋志姣
食品与发酵工业 2022年19期
李晓娇,汪玉洁,杨丽华,唐金山,宋志姣*
1(保山学院 资源环境学院,云南 保山,678000) 2(云南省高校怒江河谷生物质资源高值转化与利用重点实验室,云南 保山,678000)
果胶是一种存在于植物细胞壁和胞间层的酸性多糖[1],主链由半乳糖醛酸经α-1,4-糖苷键连接形成,侧链是由半乳糖、鼠李糖和阿拉伯糖等中性多糖组成。果胶具有良好的凝胶性、吸附性、乳化性和增稠性等特性,被广泛地应用于食品和化妆品等生产领域[2-3]。此外,果胶多糖还具有抗菌、抗氧化、调节免疫和抗癌等生物活性[4]。目前主要从柑橘皮、向日葵盘、香蕉皮和苹果皮渣中提取果胶[5-7],从小粒咖啡果皮中提取果胶还鲜有报道。
通常加工1 t小粒咖啡鲜果会产生0.5 t皮渣(包括果皮、果肉),云南小粒咖啡植区每年鲜果处理生产皮渣约39.68万t[8]。这些咖啡皮渣除部分用作堆肥外,绝大部分作为废弃物丢弃,造成环境污染和资源浪费。近年来,已有将小粒咖啡果皮制成咖啡果皮茶、果汁酒等产品,但均是小批次试验,未形成规模化生产。课题组前期研究发现,小粒咖啡果皮中果胶含量约为15%,其果胶水解物有较好的抑菌活性[9]。目前,果胶提取方法多达十余种,其中微波辅助法和超声波辅助法提取可以缩短果胶的提取时间、节约溶剂,且较大限度地保留原料的天然活性,保护果胶的组分结构[10-11]。然而,不同提取方法对果胶的理化性质和生物活性的影响不同,马丽苹等[12]通过酸碱法和高压蒸汽法改性苹果果胶,改性后的苹果果胶具有更好的抗氧化活性,因此,本研究采用盐酸浸提法(traditional acid extraction, TAE)、微波辅助法(microwae assisted extraction, MAE)和超声波辅助法(ultrasound assisted extraction, UAE)提取小粒咖啡果皮中的果胶,再通过酸碱改性和热改性获得2种改性果胶,并在分析改性前后果胶理化性质的基础上,进一步测定改性前后果胶对DPPH自由基、·OH、·O2-和ABTS阳离子自由基的清除能力,为小粒咖啡果皮资源的综合开发利用提供实验依据。
1 材料与方法
1.1 材料与仪器
小粒咖啡(CoffeaarabicaL.)果皮由云南保山老基地咖啡有限公司提供;无水乙醇、盐酸、水杨酸、硫酸亚铁、氢氧化钠、三氟乙酸,分析纯,重庆川东化工有限公司;间羟联苯、邻苯三酚,分析纯,国药集团化学试剂有限公司;聚环氧乙烷(poly ethylene oxide, PEO)、聚乙二醇(poly ethylene glycol, PEG)、岩藻糖(Fuc)鼠李糖(Rha)、阿拉伯糖(Ara)、半乳糖(Gal)、葡萄糖(Glc)、木糖(Xyl)、甘露糖(Man)、核糖(Rib)、半乳糖醛酸(GalA)、葡萄糖醛酸(GlcA),美国Sigma公司;PEO/PEG(分子质量:202 400,134 300,72 750,31 440,3 870,615 Da),安捷伦;DPPH、ABTS,国药集团化学试剂有限公司(沃凯)。
U2600紫外-可见分光光度计,日本岛津公司;PL-GPC50高效凝胶渗透色谱,美国Agilent公司;ICS5000离子色谱、Nicolet is5FTIR红外光谱仪,美国赛默飞公司;LDZX-75KB立式压力蒸汽灭菌锅,上海申安医疗器械厂;N-1001D旋转蒸发仪,东京理化公司;Noa-Nano450扫描电镜,美国FEI公司;550脱壳机,河南伍德机械设备有限公司;DZF 602真空干燥机,上海一恒科学仪器有限公司;XM-500UF智能静音超声仪,小美超声仪器(昆山)有限公司;G80F23CN3L-C2微波炉,广州电器制造有限公司。
1.2 实验方法
1.2.1 果胶提取方法
1.2.1.1 小粒咖啡果皮样品处理
小粒咖啡鲜果→[料液比:1∶8.5(g∶mL),25 ℃]水浸泡12 h→脱壳机脱壳→果皮放置通风处,太阳光下自然晾晒2~3 d直至水分含量为16.52%→粉碎机粉碎(20 000 r/min,3 min)→过80目筛→果皮粉末→加水(15 mL/g)煮沸10 min使酶失活→加入80%乙醇溶液脱色(10 min/次,重复3次)→干燥,粉碎备用。
1.2.1.2 TAE法
称取10.00 g上述小粒咖啡果皮样品于250 mL锥形瓶内,按料液比1∶25 (g∶mL)加入pH 1.5的盐酸溶液(0.032 mol/L),于60 ℃水浴中提取90 min,冷却后抽滤,滤液于40 ℃减压浓缩至原体积的1/3,冰水浴冷却后加入1.5倍体积的无水乙醇进行醇析,减压过滤,用75%乙醇洗涤,除去醇溶性杂质,滤渣于40 ℃下真空干燥至恒重,得果胶固体,称重计算得率[9]。
1.2.1.3 MAE法
称取10.00 g小粒咖啡果皮样品于250 mL锥形瓶内,按料液比1∶20 (g∶mL)加入pH 1.5的盐酸溶液,于微波功率700 W下提取5 min,冷却后抽滤,滤液按照1.2.1.2所述方法获得果胶计算得率。
1.2.1.4 UAE法
称取10.00 g小粒咖啡果皮样品于250 mL锥形瓶内,按料液比1∶20 (g∶mL)加入pH 1.5的盐酸溶液,在超声功率为200 W,温度为40 ℃的条件下提取35 min。冷却后抽滤,滤液按照1.2.1.2所述方法获得果胶计算得率。
1.2.2 小粒咖啡果皮果胶改性方法
1.2.2.1 酸碱改性
称取1.50 g小粒咖啡果皮果胶于250 mL锥形瓶中,加入100 mL、55 ℃的蒸馏水,配制得到质量浓度为15 mg/mL的果胶样品溶液。待果胶全部溶解后,缓慢加入3 mol/L NaOH溶液至pH 10.0,并置于55 ℃水浴中45 min。于室温下,加入3 mol/L HCl溶液调节pH值至3.0,在室温下静置过夜(12 h)。然后加入3 倍体积的无水乙醇,搅拌产生大量的白色絮状沉淀,过滤,滤渣于50 ℃真空烘干得到酸碱改性小粒咖啡果皮果胶[12]。改性果胶得率按公式(1)计算。
(1)
式中:m0,小粒咖啡果皮果胶质量,g;m1,改性小粒咖啡果皮果胶质量,g。
1.2.2.2 热改性
称取1.50 g小粒咖啡果皮果胶于250 mL锥形瓶中,加入50 mL、55 ℃的蒸馏水,配制得到质量浓度为30 mg/mL的果胶样品溶液。待果胶溶解后,调节pH值至4.0,置于高压蒸汽灭菌锅中,在115 ℃下维持45 min,取出冷却至室温,50 ℃下减压蒸馏至原液体积的1/3,然后向浓缩液中加入3倍体积无水乙醇,搅拌产生白色絮状沉淀,过滤,滤渣于50 ℃真空烘干得到热改性小粒咖啡果皮果胶[12]。
1.2.3 小粒咖啡果皮果胶理化性质测定
1.2.3.1 酯化度测定
称取0.10 g果胶,充分溶解于100 mL蒸馏水中。加入2~3滴酚酞指示剂,用浓度为0.10 mol/L的NaOH溶液标定,当溶液变为粉红色并保持1 min不变色时,记录所消耗的NaOH溶液体积(1,mL),即为初始滴定度。再加入浓度为3.00 mol/L的NaOH溶液2.5 mL,摇匀,5 min后加入浓度为3.00 mol/L的HCl溶液2.5 mL,振荡直至粉红色消失。然后再加入2~3 滴酚酞指示剂,用0.10 mol/L NaOH溶液滴定至再次呈粉红色并保持1 min不变色,记录NaOH溶液的消耗体积(2,mL),根据公式(2)计算酯化度。
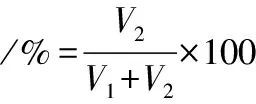
(2)
1.2.3.2 微观结构测定
将果胶样品粘于导电胶片上,喷金后在加速电压为20 k的条件下利用扫描电子显微镜观察9种果胶的表面形貌。
1.2.3.3 高效凝胶渗透色谱法(high performance gel permeation chromatography,HPGPC)测定分子质量
准确称取(10±0.05)mg果胶样品和标准品于20 mL样瓶中,向其中分别加入10 mL的流动相(水+0.2 mol/L NaNO3+0.01 mol/L NaH2PO4, pH 7.0)制成2 mg/mL溶液,加热搅拌溶解,待样品完全溶解后,微孔滤膜(0.22 μm)过滤,然后转移至2 mL进样瓶。以不同分子质量的PEO/PEG标准品作对照,制备标准曲线,测定样品的分子质量。测定条件:温度40 ℃,溶剂流速1.0 mL/min,流动相:水+0.2 mol/L NaNO3+0.01 mol/L NaH2PO4(pH 7.0),标样:PEO/PEG,进样体积100 μL;色谱柱:2× PLgel 8 μm aquagel-OH Mixed-M 7.5 mm×300 mm,检测器:示差折光检测器。
1.2.3.4 单糖组分测定
取干净的色谱瓶,精确称量多糖样品(5±0.05)mg,加入1 mL 2 mol/L三氟乙酸溶液,105 ℃加热6 h,通N2吹干。加入甲醇清洗,通氮气吹干,重复清洗2~3次。加入无菌水溶解,转入色谱瓶中待测。
将上述乙酰化产物采用DionexTMCarboPacTMPA20(150 mm×3.0 mm,10 μm)液相色谱柱测定;进样量为5 μL;流动相A(0.1 mol/L NaOH),流动相B(0.1 mol/L NaOH和0.2 mol/L NaAc),流速0.5 mL/min;柱温30 ℃;洗脱梯度:0 min (A相)∶(B相)=95∶5,30 min (A相)∶(B相)=80∶20,30.1 min (A相)∶(B相)=60∶40,45 min (A相)∶(B相)=60∶40,45.1 min (A相)∶(B相)=95∶5,60 min (A相)∶(B相)=95∶5。
1.2.3.5 傅里叶变换红外光谱(Fourier transform infrared spectroscopy,FTIR)测定
用KBr压片法测定小粒咖啡果皮果胶的红外光谱。将果胶样品干燥至恒重,按照1∶100的质量比取少量果胶与KBr混合均匀后压片,在红外光区为400~4 000 cm-1用FTIR扫描16次,扫描分辨率为4 cm-1。
1.2.4 小粒咖啡果皮果胶的抗氧化活性测定
1.2.4.1 DPPH自由基清除能力测定
取2.0 mL 0.2 mmol/L新制的DPPH溶液(95%乙醇溶解)分别与2.0 mL不同质量浓度的小粒咖啡果皮果胶样品(0.2~3.2 mg/mL)充分混匀,在室温下避光反应20 min后,在517 nm处测定吸光度,记为A1,以95%乙醇溶液代替上述体系中的DPPH溶液,在517 nm处测定吸光度,记为A2,以蒸馏水代替上述体系中果胶样品溶液,在517 nm处测定吸光度,记为A0,按公式(3)计算DPPH自由基清除率。
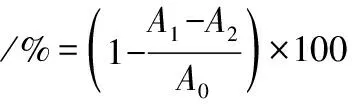
(3)
1.2.4.2 ·OH的清除能力测定
取0.6 mL 6 mmol/L FeSO4溶液分别与2.0 mL不同质量浓度的果胶样品溶液(0.2~3.2 mg/mL)混匀后,再加入0.6 mL 6 mmol/L的H2O2溶液,充分混匀后于室温下反应10 min,再分别加入6 mmol/L水杨酸0.6 mL混匀,继续反应10 min,在510 nm处测得吸光度为A1。在上述反应体系中,以无水乙醇代替水杨酸,测定吸光度为A2,以蒸馏水代替样品溶液,测定吸光度为A0,按1.2.4.1中的公式(3)计算清除率。
1.2.4.3 ·O2-清除能力测定
取不同质量浓度(0.2~3.2 mg/mL)的果胶样品溶液0.8 mL,分别加入2 mL Tris-HCl缓冲液(16 mmol/L pH 8.2)混合均匀,在25 ℃水浴30 min后,加入20 ℃水浴的邻苯三酚样品溶液0.2 mL混匀,室温下静置35 min,测定混合液在325 nm处的吸光度A1。在上述反应体系中,以蒸馏水代替邻苯三酚样品溶液测得A2,以蒸馏水代替样品测得的吸光度为A0,按公式(3)计算·O2-的清除率。
1.2.4.4 ABTS阳离子自由基清除能力测定
用磷酸盐缓冲液(pH 7.4、0.2 mol/L)配制7.4 mmol/L的ABTS溶液,加入2.6 mmol/L的过硫酸钾,室温下避光反应16 h,然后再用上述磷酸盐缓冲液稀释,使溶液在734 nm处的吸光度A0为(0.70±0.02),得ABTS阳离子自由基稀释液。将果胶样品用磷酸盐缓冲液配制成质量浓度为 0.2、0.4、0.8、1.6、3.2 mg/mL的溶液,分别量取0.2 mL不同质量浓度果胶样品溶液与2.0 mL ABTS阳离子自由基稀释液混合,摇匀,室温避光反应20 min后,在734 nm处测定吸光度(A1);同时将0.2 mL不同质量浓度的果胶溶液与2 mL 磷酸盐缓冲液振摇20 s使其混合,并室温避光反应20 min以后,在734 nm处测定吸光度(A2)。按公式(3)计算ABTS阳离子自由基清除率。
1.3 数据处理
所有试验均进行3次重复试验。采用Origin 2019进行作图,SPSS 22.0进行多重比较分析,显著水平为0.05。
2 结果与分析
2.1 提取方法对果胶得率的影响
3种不同方法提取的果胶得率如图1所示,得率大小为:MAE法(7.24%)>UAE法(6.88%)>TAE法(5.21%)。通过多重比较分析,MAE法和UAE法的果胶得率无显著差异(P>0.05),而TAE法果胶得率较MAE和UAE法差异显著(P<0.05)。可能原因是MAE法的热效应和UAE法的空化效应和机械效应对小粒咖啡果皮的细胞壁的分解破坏程度大,使细胞更容易破裂,从而细胞中更多的多糖等物质被分解出来,所得果胶得率相对较高[13]。

图1 不同提取方法的果胶得率Fig.1 Pectin yield of different extraction methods
2.2 改性小粒咖啡果皮果胶的理化性质
3种方法提取的果胶分别经过酸碱改性和热改性共得到6种不同的改性果胶,其理化性质见表1。TAE法热改性果胶(A3)产率36.50%,略高于酸碱改性果胶(A2)产率35.69%;MAE法的2种改性果胶产率均达到40%以上,高于其他2种方法所得的改性果胶;UAE法的2种改性果胶产率最低,为28.44%和28.69%。与改性前相比,6种改性果胶的GalA含量均显著提高(P<0.05)。改性前3种方法提取的果胶酯化度均高于30%,而改性后酯化度均显著降低(P<0.05),且均降低到25%以下,且酸碱改性的果胶酯化度最低。果胶在酸碱改性时,碱处理产生β-消除反应能使同聚半乳糖醛酸骨架解聚,同时也使酯化度降低;酸处理则优先脱下侧链上的中性糖[12,14],因此,果胶通过酸碱改性后,其GalA含量增加,而酯化度下降;果胶热改性,高温高压会引起β-消除反应和脱甲酯基作用[15],从而也导致酯化度下降,GalA含量增加。
式中,W′维数为s×m,θ2为阈值,维数为m×1,f2(I2)为输出层的传递函数,可采用Sigmoid函数或Purelin函数。
如表1所示,9种果胶的阿拉伯糖(Ara)、半乳糖(Gal)均含量相对较高,其次是鼠李糖(Rha)、和葡萄糖(Glc)。改性前果胶的中性糖含量均高于改性后果胶,且TAE法提取的果胶中性糖含量最高。改性后果胶的糖醛酸含量均高于改性前的果胶,且MAE法提取的果胶改性前后的糖醛酸含量均高于TAE法和UAE法提取的果胶。以Rha与GalA相对含量比表示果胶的分支程度,比值越高说明分支度更高[16]。表1中Rha/GalA的比值为0.10~0.30,表明不同提取方式和改性方法得到的9种果胶有较多支链,且果胶局部降解较少,其结构主要为RG-I型[17]。
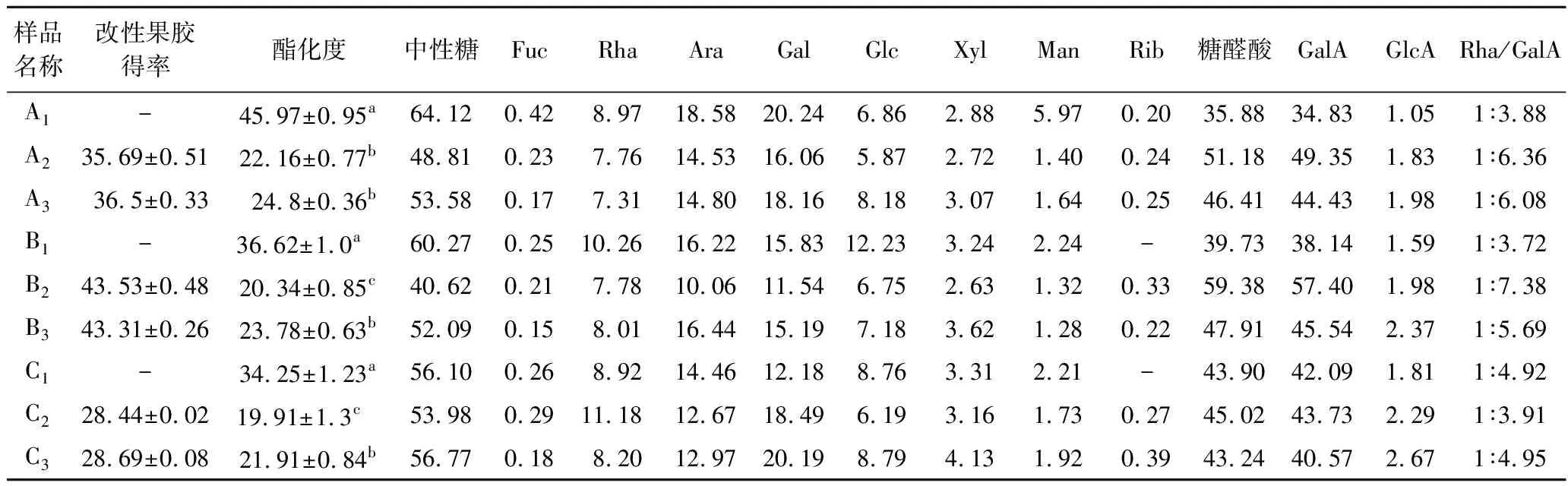
表1 改性前后小粒咖啡果皮果胶得率和理化性质 单位:%
2.3 小粒咖啡果皮果胶的扫描电镜分析
图2是改性前后小粒咖啡果皮果胶的扫描电子显微镜图,未改性的A1、B1和C1果胶表面凹凸不平但质地比改性后的果胶紧密,其中C1果胶表面凸起较多,并且有特殊的折叠结构和少量的孔隙;A1和B1果胶质地紧密无明显孔隙结构。酸碱改性果胶(A2、B2、C2)质地都较为疏松,其中B2果胶表面有颗粒状堆积,C2果胶存在空腔结构,而A2果胶无明显颗粒堆积和孔隙结构。热改性果胶(A3、B3、C3)与未改性和酸碱改性果胶相比表面质地更加疏松,存在空腔结构或片层结构。不同的提取方法及改性方法使得果胶的分子质量、多糖含量、酯化度等都发生了不同程度的变化,从而导致果胶多糖存在结构形态上的差异。
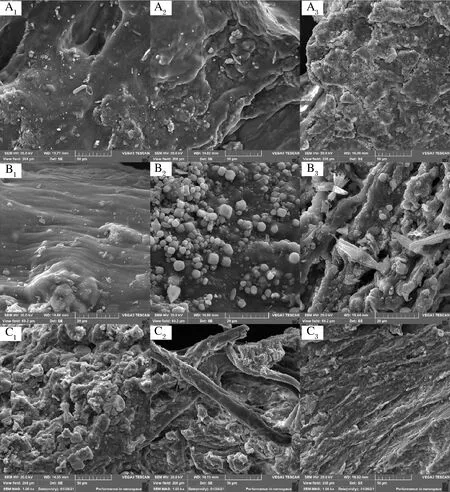
图2 改性前后小粒咖啡果皮果胶的扫描电子显微镜图Fig.2 Scanning electron micrograph of natie and modified C.arabica pomace pectin
2.4 不同提取方式和改性对分子质量的影响
利用HPGPC法对改性前后的果胶分子质量进行测定,结果如表2所示。改性前后的果胶分子质量在1.42×102~6.79×104Da,不同提取方式和改性方法制备的小粒咖啡果皮果胶多糖分子质量分布不均一,均由3种不同分子质量的组分峰组成(峰1,峰2,峰3)。9种果胶的峰2和峰3的分散系数(Mw/Mn)均接近1.0,说明分子质量分布比较集中,峰1的分散系数(Mw/Mn)均大于2.0,分布比较分散。比较这9种果胶峰面积较大的峰(即峰1)的重均分子质量:A1>A3>A2,B1>B2>B3,C1>C2>C3,而A3、B3、C3这部分的果胶含量相对较高(峰面积较大);比较峰2的重均分子质量:A1>A3>A2,B1>B3>B2,C1>C3>C2,而A3、B2、C2这部分的果胶含量相对较高;比较峰3的重均分子质量:A1>A3>A2,B1>B2>B3,C1>C2>C3,而A1、B1、C2这部分的果胶含量相对较少。其中酸碱改性和热改性的重均分子质量均小于未改性果胶的分子质量,这可能是由于酸碱和高压加热的条件下,果胶分子发生β-消除反应和去酯化反应,使得果胶主链断裂,生成一些聚半乳糖醛酸低聚物[18]。

表2 改性前后小粒咖啡果皮果胶的分子质量Table 2 The relatie molecular weight and its distributionof natie and modified C. arabica pomace pectin
续表2
2.5 小粒咖啡果皮果胶的红外光谱分析


图3 改性前后的小粒咖啡果皮果胶的红外光谱图Fig.3 The FTIR of natie and modified C.arabica pomace pectin
2.6 改性小粒咖啡果皮果胶的抗氧化活性
2.6.1 DPPH自由基清除能力
由图4可知,随着果胶质量浓度的增加,9种不同果胶对DPPH自由基的清除率也不同程度地增加。当果胶质量浓度为0.2 mg/mL时,3种未改性的果胶A1、B1、C1对DPPH自由基的清除率均低于酸碱改性和热改性的果胶;当果胶质量浓度大于0.8 mg/mL时,酸碱改性的果胶A2、B2、C2对DPPH自由基的清除率均高于未改性和热改性的果胶,而A1和A3,B1和B3之间对DPPH自由基的清除率无显著差异。当果胶样品质量浓度为6.2 mg/mL时,9种果胶对DPPH自由基的清除率均大于90%,其中酸碱改性果胶A2、B2、C2对DPPH自由基的清除率最高,分别为97.36%、99.12%和98.11%,IC50值分别为0.58、0.52、0.53 mg/mL(表3),高于南酸枣果胶多糖(IC50=3.08 mg/mL)[22]。可见酸碱改性的小粒咖啡果皮果胶具有更好的清除DPPH自由基的能力。A2、B2、C2果胶的GalA含量均高于未改性和热改性的果胶,因此具有更强的抗氧化活性。
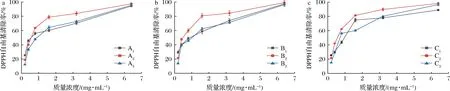
a-TAE法果胶;b-MAE法果胶;c-UAE法果胶图4 不同小粒咖啡果皮果胶对DPPH自由基的清除率Fig.4 Scaenging capacity of different C.arabica pomace pectins against DPPH radical

表3 改性前后小粒咖啡果皮果胶清除4种自由基的IC50值Table 3 The IC50 alues of natie and modified C.arabica pomace pectin
2.6.2 ·OH的清除能力
由图5可知,随着果胶质量浓度的增加,9种果胶对·OH的清除率也有不同程度的增加。当果胶质量浓度为0.02~6.4 mg/mL时,A2果胶对·OH的清除率要高于A1和A3,果胶质量浓度小于0.08 mg/mL时,A3果胶低于A1果胶。当果胶质量浓度高于1.6 mg/mL时,对·OH的清除率由高到低依次为:A2>A3>A1。果胶质量浓度为0.02~6.4 mg/mL时,对·OH的清除率由高到低依次为:B2>B3>B1,当果胶质量浓度为0.08 mg/mL时,B1和B3对·OH的清除能力无显著差异。在果胶质量浓度为0.08 mg/mL时,B1和B3对·OH的清除率分别为41.20%和41.64%,无显著差异,而B2果胶的清除率则小于前两者,仅为32.54%。当果胶质量浓度大于1.6 mg/mL时,对·OH的清除率由高到低依次为:C2>C3>C1。
当果胶质量浓度为6.4 mg/mL时,9种小粒咖啡果皮果胶(A1~A3,B1~B3,C1~C3)对·OH的清除率均大于90%,其中TAE法提取和改性得到的A1、A2、A33种果胶均大于96%,3种提取方法中,均为酸碱改性的果胶A2、B2、C2的清除率最高,分别为:99.97%、98.56%和99.23%,IC50值分别为0.77、0.68、0.78 mg/mL(表3),高于南酸枣果胶多糖(IC50=3.69 mg/mL)。果胶多糖中的羧基可以络合Fe2+,从而抑制Fe2+与H2O2生成·OH,同时果胶多糖中的羧基和羟基可作为氢供体与·OH反应[23-25]。而A2、B2、C2中的GalA含量相对较高(表1),由此可知,GalA相对含量较高的酸碱改性小粒咖啡果皮果胶对·OH有更强的清除能力。

a-TAE法果胶;b-MAE法果胶;c-UAE法果胶图5 不同小粒咖啡果皮果胶对·OH的清除率Fig.5 Scaenging capacity of different C.arabica pomace pectins against ·OH
2.6.3 ·O2-的清除能力
由图6可知,9种果胶均随着果胶质量浓度的增加·O2-的清除率也在不同程度的增加。当果胶质量浓度为1.6 mg/mL时,热改性(A3)和未改性果胶(A1)对·O2-的清除能力无显著差异。质量浓度小于1.6 mg/mL时,果胶对·O2-的清除率为A2>A1>A2,而当果胶质量浓度高于1.6 mg/mL时,对·O2-的清除率为A2>A3>A1。在果胶浓度为1.6~6.4 mg/mL时,对·O2-的清除率为B2>B3>B1,当果胶质量浓度小于1.6 mg/mL时,清除能力为B3>B2>B1。C1和C3在质量浓度在0.2~0.8 mg/mL时,对·O2-的清除率无显著差异。当果胶质量浓度高于0.8 mg/mL时清除率为C2>C3>C1。
当果胶质量浓度为6.4 mg/mL时,9种小粒咖啡果皮果胶(A1~A3,B1~B3,C1~C3)对·O2-的清除率均大于90%,3种提取方法中,酸碱改性的果胶A2、B2、C2的清除率最高,分别为99.87%、97.28%和99.92%,IC50值分别为0.49、0.68、0.54 mg/mL(表3),由于UAE法提取的果胶GalA含量最高,因此对·O2-的清除能力高于TAE和MAE法提取的果胶。

a-TAE法果胶;b-MAE法果胶;c-UAE法果胶图6 小粒咖啡果皮果胶对·O2-的清除率Fig.6 Scaenging capacity of different C.arabica pomace pectins against ·O2-
2.6.4 ABTS阳离子自由基的清除能力
由图7可知,随着果胶质量浓度的增加9种果胶对ABTS阳离子自由基的清除率也随之增加。在果胶质量浓度为0.2~0.4 mg/mL时,A1、A2和A3对ABTS阳离子自由基的清除率的差异不显著,当果胶质量浓度高于0.8 mg/mL时,呈现A2>A3>A1的规律。在果胶质量浓度高于1.6 mg/mL时,对ABTS阳离子自由基的清除率呈现B2>B3>C1的规律。果胶质量浓度低于1.6 mg/mL时,B2对ABTS阳离子自由基的清除率均小于B1和B3,B1和B3无显著差异。呈现B2>B3>B1的规律,当果胶质量浓度为6.4 mg/mL时,9种小粒咖啡果皮果胶(A1~A3, B1~B3, C1~C3)对ABTS阳离子自由基的清除率均大于90%,其中MAE法和UAE法提取的果胶(B1~B3, C1~C3)清除率均大于96%,3种提取方法中,酸碱改性的果胶A2、B2、C2的清除率最高,分别为99.12%、99.97%和99.82%,IC50值分别为0.76、0.89、0.90 mg/mL(表3)。
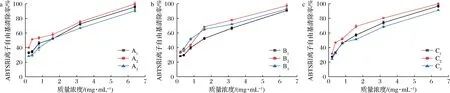
a-TAE法果胶;b-MAE法果胶;c-UAE法果胶图7 小粒咖啡果皮果胶对ABTS阳离子自由基的清除率Fig.7 Scaenging capacity of different C.arabica pomace pectins against ABTS cationicradical
3 结论与讨论
本研究采用TAE、MAE和UAE 3种方法提取小粒咖啡果皮中的果胶,其中MAE法果胶得率最高,3种方法提取的果胶的半乳糖醛酸含量为C1>B1>A1,酯化度为A1>B1>C1;改性前后9种果胶(A1~A3, B1~B3,C1~C3)的阿拉伯糖(Ara)、半乳糖(Gal)均相对较高。果胶酸碱改性和热改性后,3种方法提取的果胶的GalA含量均升高,酯化度均降低。2种改性方法中,酸碱改性的果胶的GalA含量高于热改性果胶,MAE法酸碱改性后的果胶(B2)GalA含量最高。改性果胶质地都较为疏松、有空腔结构或片层结构。改性前后的果胶分子质量在5.62×102~6.79×104Da,均由3种不同分子质量的组分峰组成。不同的提取方法及改性方法使得果胶的分子质量、多糖含量、酯化度等都发生了不同程度的变化,从而导致果胶多糖存在结构形态上的差异。
9种果胶对DPPH自由基、·OH、·O2-和ABTS阳离子自由基的清除率呈现质量浓度依赖效应,其中当果胶质量浓度大于3.2 mg/mL时,清除能力均为酸碱改性果胶>热改性果胶>未改性果胶。超声波辅助提取和酸碱改性果胶的半乳糖醛酸含量较高,且对4种自由基的清除能力也相对较好,因此果胶中半乳糖醛酸的含量和对自由基的清除能力存在正比关系。本研究为以小粒咖啡果皮为原料的天然食源性抗氧化剂的开发提供了理论基础和科学依据,具有潜在的经济效益和社会效益。
猜你喜欢
作物学报(2022年2期)2022-11-06
农业工程学报(2022年14期)2022-10-19
现代仪器与医疗(2022年4期)2022-10-08
食品科学(2022年12期)2022-07-07
农产品加工(2022年9期)2022-06-17
科普童话·神秘大侦探(2021年12期)2021-01-19
课堂内外(高中版)(2021年9期)2021-01-17
大灰狼(2020年3期)2020-04-24
天津农业科学(2017年7期)2017-07-25
中国实用医药(2016年26期)2016-11-07
