
The cuproptosis-related gene signature predicts clinical diagnosis, prognosis, and immune microenvironment for glioblastoma multiforme
2022-10-13 13:53:28GangLiuLiWangZhenZhenPeiJiZhouYangBoWenDengShengYuanJiangRuiQinYuHuiZhongBaiLinXuXiaoHongMu
Precision Medicine Research 2022年3期
Gang Liu,Li Wang,Zhen-Zhen Pei,Ji-Zhou Yang,Bo-Wen Deng,Sheng-Yuan Jiang,Rui-Qin Yu,Hui-Zhong Bai,Lin Xu*,Xiao-Hong Mu*
1Department of Orthopedics, Dongzhimen Hospital, Beijing University of Chinese Medicine, Beijing 100010, China. 2Beijing An Yuan Quan Lv Medical Research Institute,Beijing 100020,China. 3Department of Neurosurgery,Dongfang Hospital,Beijing University of Chinese Medicine,Beijing 100071,China.
Abstract
Keywords: cuproptosis; glioblastoma multiforme; prognosis; immune microenvironment;drug sensitivity
Background
Glioblastoma multiforme (GBM) is the most common primary malignant tumor of the brain and stands out as the most malignant one of all astrocytic tumors, accounting for 57% of all gliomas and 48% of primary malignant central nervous system (CNS) tumors [1].Although there has been significant progress in the treatment of GBM,which primarily consists of surgery, radiotherapy, chemotherapy, and targeted therapy, the generally low survival rate and propensity to return after surgery remain significant problems that clinicians must urgently address. GBM poses a substantial threat to the health of patients, causes irreparable harm to families, and places a significant financial strain on society [2]. To change the status quo of GBM treatment, it is essential to explore future immunotherapies and precision oncology approaches. The purpose of this work was to explore an effective prognostic model to predict prognosis and immunotherapeutic response and to identify specific drugs with high sensitivity against GBM.
Cuproptosis is a novel way of cell death, and its main process depends on the accumulation of intracellular copper ions in cells. The lipid acylated components of the tricarboxylic acid cycle(TCA)can be combined directly with copper ions, resulting in accumulation and dysregulation of the TCA, triggering proteotoxic stress and cell death[3].
Some cuproptosis-related genes (CRGs) are closely associated with gliomas. NFE2L2 has been revealed as a potential prognostic biomarker associated with immune infiltration in low-grade glioma(LGG) [4]. NLRP3 promotes the growth and invasion of gliomas via the IL-1β/NF-κB p65 signals [5]. ATP7B limits GBM’s resistance to temozolomide, whose high expression reduces overall survival (OS)[6]. Inhibition of SLC31A1 retards the progression of glioma cell growth [7]. Although cuproptosis-related genes (CRGs) are closely associated with the development and progression of GBM, whether these genes have a prognostic role for GBM and the molecular functions involved are not clear. Systematic studies on the role of CRGs in GBM remain insufficient.
We integrated data from The Cancer Genome Atlas (TCGA)databases and the public Gene Expression Omnibus (GEO) to clarify the prognostic role of CRGs in patients with GBM,explore the immune microenvironment of different risk types of GBM and search for highly sensitive drugs. In GBM, we identified three different CRGs clusters,obtained common differential genes, and performed KEGG and GO analyses. Two key genes related to prognosis were revealed. Finally, a risk-scoring model based on CRGs was constructed to predict clinical diagnosis,prognosis,immune microenvironment,and drug sensitivity.This study lays the foundation for further research between CRGs and GBM.
Methods
Data acquisition
Clinical data and raw RNA-seq data on GBM were downloaded from The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/) and Gene Expression Omnibus(GEO,https://www.ncbi.nlm.nih.gov/geo).174 GBM samples were obtained from the TCGA database. 50 GBM samples were obtained from the GEO database. Data on copy number variants (CNV) and somatic mutations for all genes were acquired from the UCSC Xena browser (https://xenabrowser.net). The chi-square test was used to determine the CNV differences for all genes(p< 0.05). The RCircos R package demonstrated where the significantly different genes were located on the chromosomes.
Identification of CRGs and differentially expressed genes (DEGs)between GBM and adjacent nontumor tissues
According to the available literature, which includes NFE2L2, NLRP3,ATP7B, ATP7A, SLC31A1, FDX1, LIAS, LIPT1, LIPT2, DLD, DLAT,PDHA1, PDHB, MTF1, GLS, CDKN2A, DBT, GCSH, and DLST, CRGs had been collected [3, 8, 9, 10, 11, 12]. We identified tumor tissues and adjacent nontumor tissues from TCGA-GBM, and then compared the expression levels of CRGs between the two groups.The R package‘limma’ (|logFC|>1 andp<0.05) was calculated to acquire DEGs.TCGA and GEO clinical data were combined to obtain the optimal cutoff, samples were divided into high and low expression groups according to the expression levels of CRGs. Kaplan-Meier (K-M)survival curves were used to evaluate the influence of every CRGs on prognosis. Statistically significant was defined asp< 0.05.
Construction of a scoring system and calculate of the risk score
We used the ConsensusClusterPlus package in R to cluster the samples for maximum stability. According to the differences in CRGs expression levels, the samples were divided into three CRG clusters and 21 DEGs were identified.Then,the samples were divided into two Geen clusters according to the 21 DEGs expression levels.We used the‘glmnet’ package in R to conduct the lasso cox regression model and constructed a multigene signature of the cuproptosis-related DEGs in the TCGA group.The formula for computing risk score is as follows:
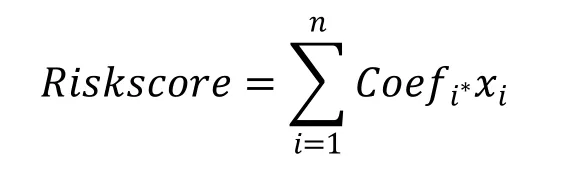
where xiis the expression value for each CRG and Coefi stands for the coefficients. Each patient with a GBM had their risk score determined using this formula. Receiver operating characteristic (ROC) and area under the curve were used to measure the prognostic signature's ability to predict clinical characteristics and survival (AUC).
We used the ‘survival’ and ‘regplot’ packages in R 4.1.3 to conduct a nomogram for prognosis prediction.
Functional enrichment analyses
The'ggplot2' package in R software version 4.1.3 was used to perform Kyoto Encyclopedia of Genes and Genomes(KEGG)pathway and Gene Ontology (GO) analysis, including the biological process(BP), cellular component (CC), and molecular function (MF), to get insight into the functional implications.
Immune-related analysis
The ‘gsva’ package in R software version 4.1.3 was used to calculate single-sample gene set enrichment analysis(ssGSEA) scores for exploring immune infiltration in GBM. We used the'estimate' package in R software version 4.1.3 to investigate the immune score, stromal score, and estimate score of TCGA-GBM, then evaluated differences between high and low-risk groups.
Drug sensitivity analysis
The pRRophetic package in R software version 4.1.3 was used to select drugs with different sensitivities in different risk groups. Statistical significance was set atp<0.05.
Statistical analysis
The R software 4.1.3 was used to examine all the data. Kaplan-Meier curves were used to compare the OS between different groups of GBM patients. Spearman's coefficient was used to measure correlations. We evaluated the difference in gene expression levels between groups via Kruskal-Wallis tests. The RCircos R program was used to visualize the landscape of CNV and the position of the genes.Statistical significance was set atp<0.05.
Results
The landscape of CRGs in GBM
This investigation includes 19 CRGs. First, we looked at how these genes were expressed in both normal tissues and TCGA-GBM tissues(Figure 1A), and the results showed that the expression levels of 10 genes were significantly different between the normal group and the tumor group. NFE2L2 ATP7A, SLC31A1 FDX1, LIPT1, CDKN2A, DLST were raised in GBM (p<0.05), ATP7B, PDHA1, GLS were reduced in GBM(p<0.05). Subsequently, CNV and chromosome location were studied. CDKN2A and ATP7B were the genes with the highest copy number loss frequency, in which CDKN2A was located on chromosome 9 and ATP7B on chromosome 13 (Figure1 B, C). Finally,we further studied the mutation frequency of CRGs in the TCGA-GBM data set, and the results showed that the mutation frequency of NLRP3, CDKN2A, and other 7 genes was greater than 1%, among which NLRP3 mutation frequency was the highest, mainly missense mutation (Figure 1D).
The relationship between CRGs and prognosis
Two RNA-seq datasets were integrated to explore the prognostic significance of CRGs, including TCGA-GBM (n= 174) and GSE83300(n= 50). Among them, 8 genes, such as DLD, FDX1, and NFE2L2 are regarded as risk factors, and 9 genes, such as DLST and GLS, are regarded as protective factors (Figure 2A). After using the Kaplan-Meier survival curve, ATP7A, DLD, LIPT2, NFE2L2, and SLC31A1 five high gene expressions were significantly associated with poor prognosis. ATP7B, CDKN2A, DBT, DLST GCSH, and MTF1 six high gene expressions were significantly associated with improved prognosis (Figure 2B-L). The prognosis of patients is directly correlated with these genes.
Classification strategy of CRGs and ssGSEA analysis in CRGclusters
According to the expression of CRGs in the samples, we conducted clustering for further study (Figure 3A). The empirical cumulative distribution function (CDF) plots showed the consensus distributions for k (2-9) (Figure 3B-C). Based on the consensus matrix, we chose k= 3. Principal component analysis (PCA) showed a good clustering difference (Figure 3D). Kaplan-Meier survival curve was used to explore the prognosis of different CRG clusters. The results showed that there were significant differences in OS among different CRG clusters (p= 0.043), there was the poorest outlook in cluster B.(Figure 3E). The ssGSEA algorithm was applied to calculate the enrichment of immune cell components in the immune microenvironment of three CRG clusters and found that more immune cells were significantly enriched in cluster B (Figure 3F). Different regulation modes based on CRG reflected the mechanism of immune infiltration.
GSVA analysis in CRGcluster
The expression and clinical signs of three CRGs clusters are shown in a heatmap (Figure 4A). We assessed the significantly enriched KEGG and GO to get insight into the functional implications via GSVA. For KEGG, compared with cluster A, linoleic acid metabolism, drug metabolism cytochrome p450, and olfactory transduction are highly expressed in cluster B. Compared with cluster C, ABC transporters,ECM receptor interaction, O-glycan biosynthesis, nicotinate,nicotinamide metabolism, and apoptosis were highly expressed in cluster B (Figure 4B-D). For GO, biological process, such as positive regulation of mitochondrial outer membrane permeabilization in apoptotic signaling pathway, and outer membrane permeabilization were enriched significantly in cluster B (Figure 5A-C).
GSVA analysis in CRGcluster
The expression and clinical signs of three CRGs clusters are shown in a heatmap (Figure 4A). We assessed the significantly enriched KEGG and GO to get insight into the functional implications via GSVA. For KEGG, compared with cluster A, linoleic acid metabolism, drug metabolism cytochrome p450, and olfactory transduction are highly expressed in cluster B. Compared with cluster C, ABC transporters,ECM receptor interaction, O-glycan biosynthesis, nicotinate,nicotinamide metabolism, and apoptosis were highly expressed in cluster B (Figure 4B-D). For GO, biological process, such as positive regulation of mitochondrial outer membrane permeabilization in apoptotic signaling pathway, and outer membrane permeabilization were enriched significantly in cluster B (Figure 5A-C).
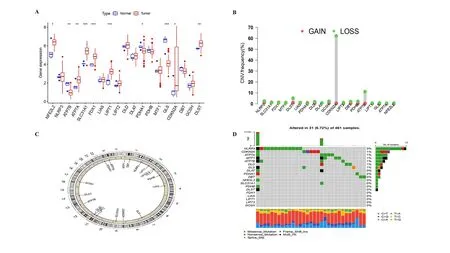
Figure 1.The landscape of CRGs in TCGA-GBM.(A)CRGs’s expression in tumor group and normal group.Genes upregulation was shown in red,genes downregulation in blue. (B) The frequency of CNV. CNV gain was shown in red, and CNV loss in green. (C) Chromosomal location of CNV.(D) The mutation of CRGs in GBM. Splicing mutations were represented in orange, frameshift mutations in blue, missense mutations in green, and nonsense mutations in red.

Figure 2. The relationship between CRGs and prognosis. (A) The degree distribution network of the prognostic-related CRGs. High-risk genes were shown in yellow circles and low-risk genes in green circles.Positive correlations between genes were represented with pink lines and negative correlations with blue lines. Correlation coefficients was set at p <0.0001. The p-value was shown with different sizes of the dot. (B-L) 11 CRGs are significantly associated with prognosis,High gene expression levels was represented in red, and low gene expression levels in blue.
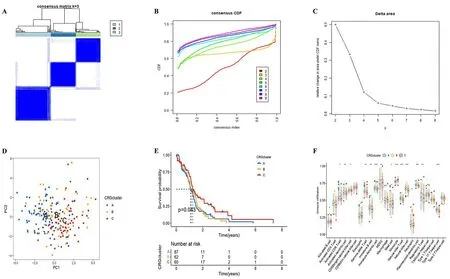
Figure 3.Classification strategy of CRGs and ssGSEA analysis in CRGclusters.(A-C)The unsupervised clustering process for CRGs. (D) A PCA plot of CRGclusters (k = 3). (E) The Kaplan-Meier survival curve showed the relationship between CRGclusters and OS (p = 0.043). (F) The box plot showed the difference in immune cell infiltration between CRGclusters using ssGSEA (*p <0.05,**p <0.01, ***p <0.001).
DEGs identify and functional enrichment analyses
To look for genes associated with specific phenotypes for each cuproptosis regulation pattern, we used LIMMA packages to identify DEGs. A total of 21 DEGs were screened out from the different CRGclusters (Figure 6A). KEGG and GO analyses were performed on 21 DEGs. The most important pathway for this DEG enrichment along the KEGG pathway was ferroptosis, porphyrin metabolism, and legionellosis. Oligodendrocyte development central nervous system myelination, axon ensheathment in the central nervous system,oligodendrocyte differentiation, glial cell development, myelination,ensheathment of neurons, axon ensheathment, regulation of myelination, and positive regulation of gliogenesis were highly enriched by GO analyses(Figure 6B-E).
Cluster analysis of DEGs
According to the expression levels of DEGs in different samples, the samples were clustered. Based on the consensus matrix, we chose k =2 (Figure 7A), The empirical cumulative distribution function (CDF)plots showed the consensus distributions for k (2-9) (Figure 7B-C).Clinical prognostic analysis of the two clusters revealed significant differences in survival (p= 0.008) (Figure 7D). 5 DEGs associated with prognosis were selected by univariate cox analysis and their expression levels were showed with heatmap (Figure 7E). To explore the relationship between CRGs and DEG,the expression levels of CRGs in the two clusters were compared, and it was found that NFE2L2 and the other 10 CRGs had significant expression differences, which were closely related to prognosis.
Construct the DEGs prognostic signature
Among the genes associated with prognosis, 5 DEGs were associated with OS in GBM patients (Figure 8A). We then performed lasso regression analysis to eliminate prognostic false-positive DEGs and further screened 4 DEGs (Figure 8B). Multivariate cox regression analysis was used to evaluate the influence of these 4 prognostic DEGs on the OS and clinical outcome of GBM patients(p<0.05).Finally, 2 genes (NEAT1 and ZNF488) with independent prognostic values were identified, which were further applied to establish prognostic risk models. According to Kaplan-Meier survival curve results, patients with a high-risk score of GBM had a poor clinical prognosis (Figure 8C-E). Next, we created ROC curves for 1, 3, and 5 years, and discovered that the survival statuses of GBM were divided effectively by the risk score. (Figure 8F,1-year AUC = 0.616,3-year AUC =0.694, 5-year AUC = 0.890). We established a nomogram model,including age, sex, and risk score, to predict patient survival probability (Figure 8G). The heatmap showed the expression of NEAT1 and ZNF488 genes in the high and low-risk groups. NEAT1 was a risk gene and ZNF488 was a protection gene (Figure 8H).
Correlation analysis of immune cells and differences in TME
The correlation between immune cells and risk score was further studied and found that neutrophils and macrophages M0 were significantly positively correlated, while monocytes and eosinophils were significantly negatively correlated (Figure 9A). The scatterplot also verified this conclusion (Figure. 9B-E). We calculated the proportion of immune matrix components in the tumor microenvironment (TME) by immune score, stromal score, and estimate score, which were positively correlated with immunity,matrix, and the sum of the two, respectively. The high-risk group's stromal score, immunological score, and estimation score were all significantly greater than those of the low-risk group, indicating that the higher the risk score,the lower the tumor purity and the worse the prognosis
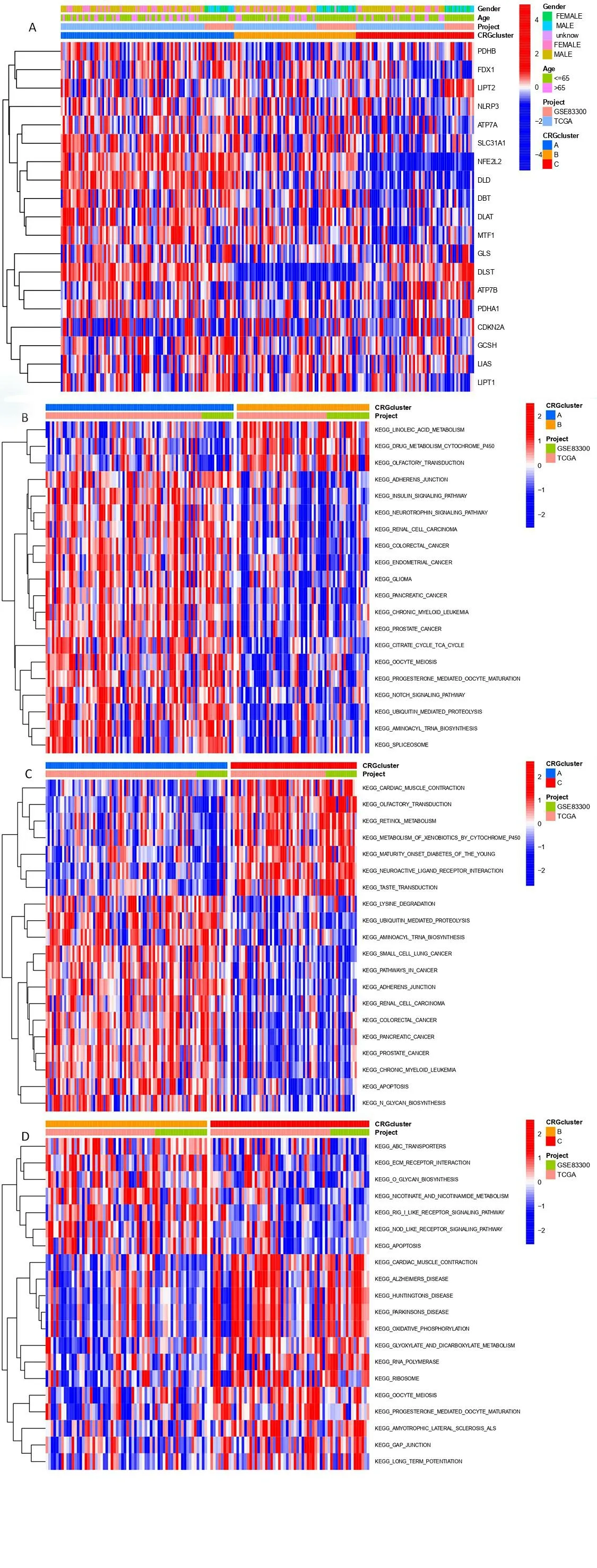
Figure 4. GSVA analysis in CRGclusters. (A) The heatmap of CRGclusters and its clinical characteristics. (B-D) Visualization of the results of KEGG with CRGclusters.
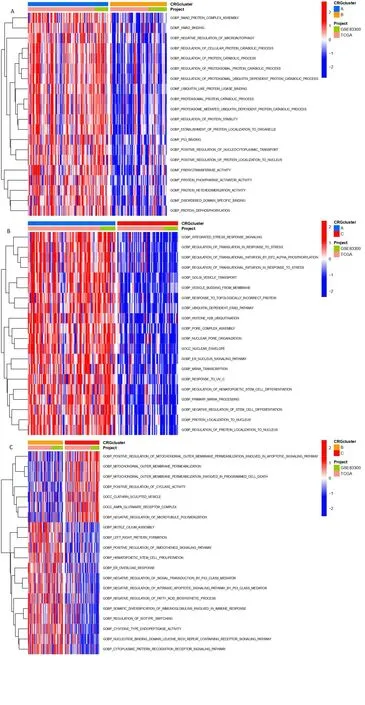
Figure 5.GSVA analysis in CRGclusters. (A-C) Visualization of the results of GO with CRGclusters.
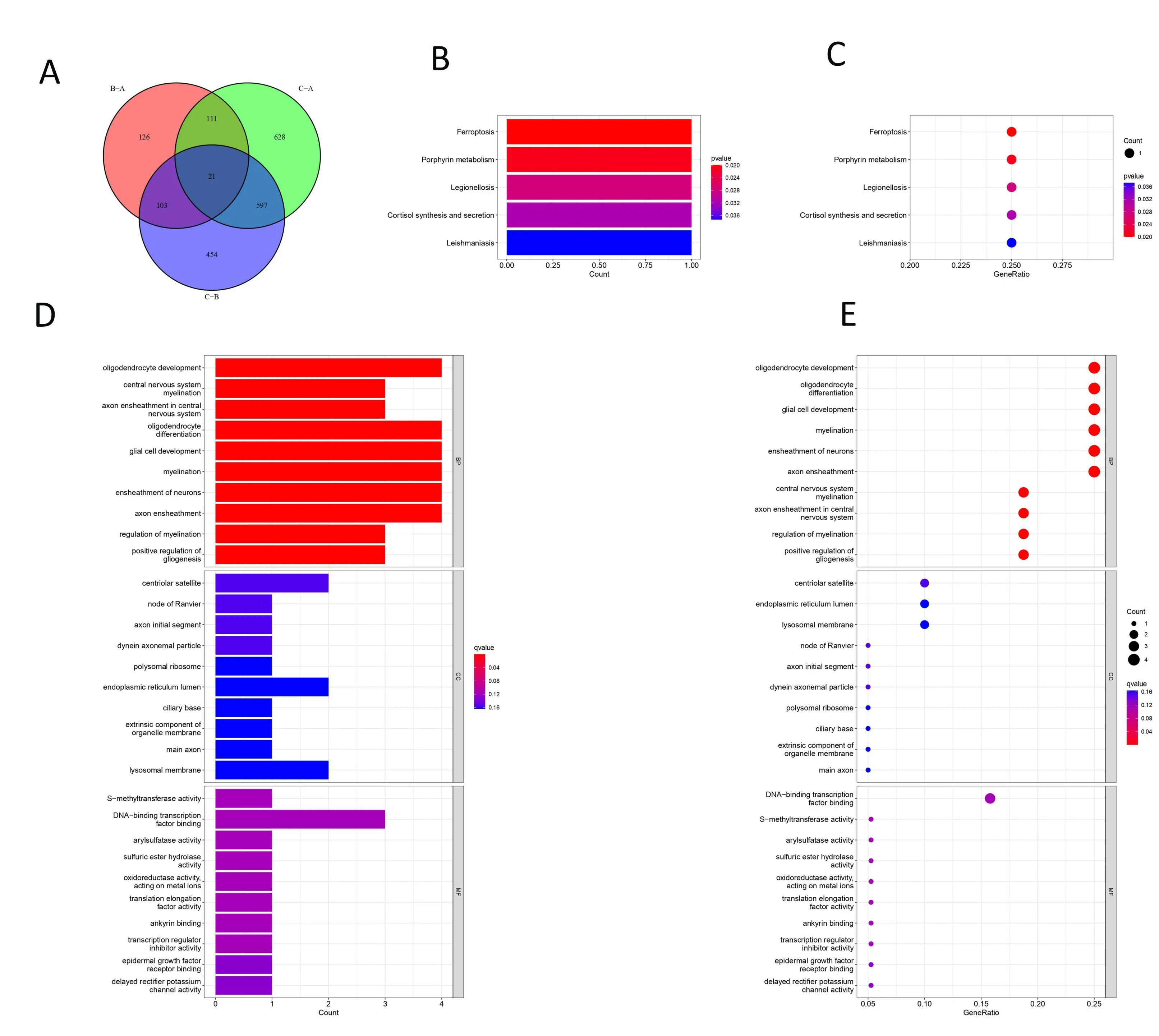
Figure 6. DEGs identify and functional enrichment analyses (A) A Venn diagram of DEGs in CRGclusters (p = 0.001). (B-C) GO analyses of DEGs. (D-E) KEGG pathway analyses of DEGs.
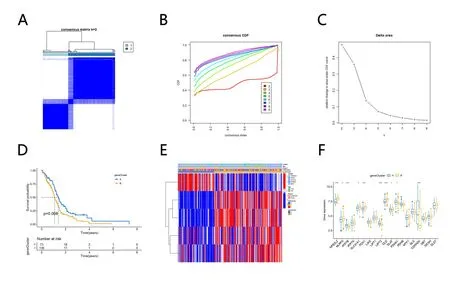
Figure 7. Cluster Analysis of DEG Levels. (A-C) The unsupervised clustering process for DEGs. (D) The Kaplan-Meier survival curve showed the relationship between CRGclusters and OS (p=0.008).(E) The heatmap of different geneclusters. (F) The expression levels of CRGs in geneclusters(*p <0.05, **p <0.01,***p <0.001).
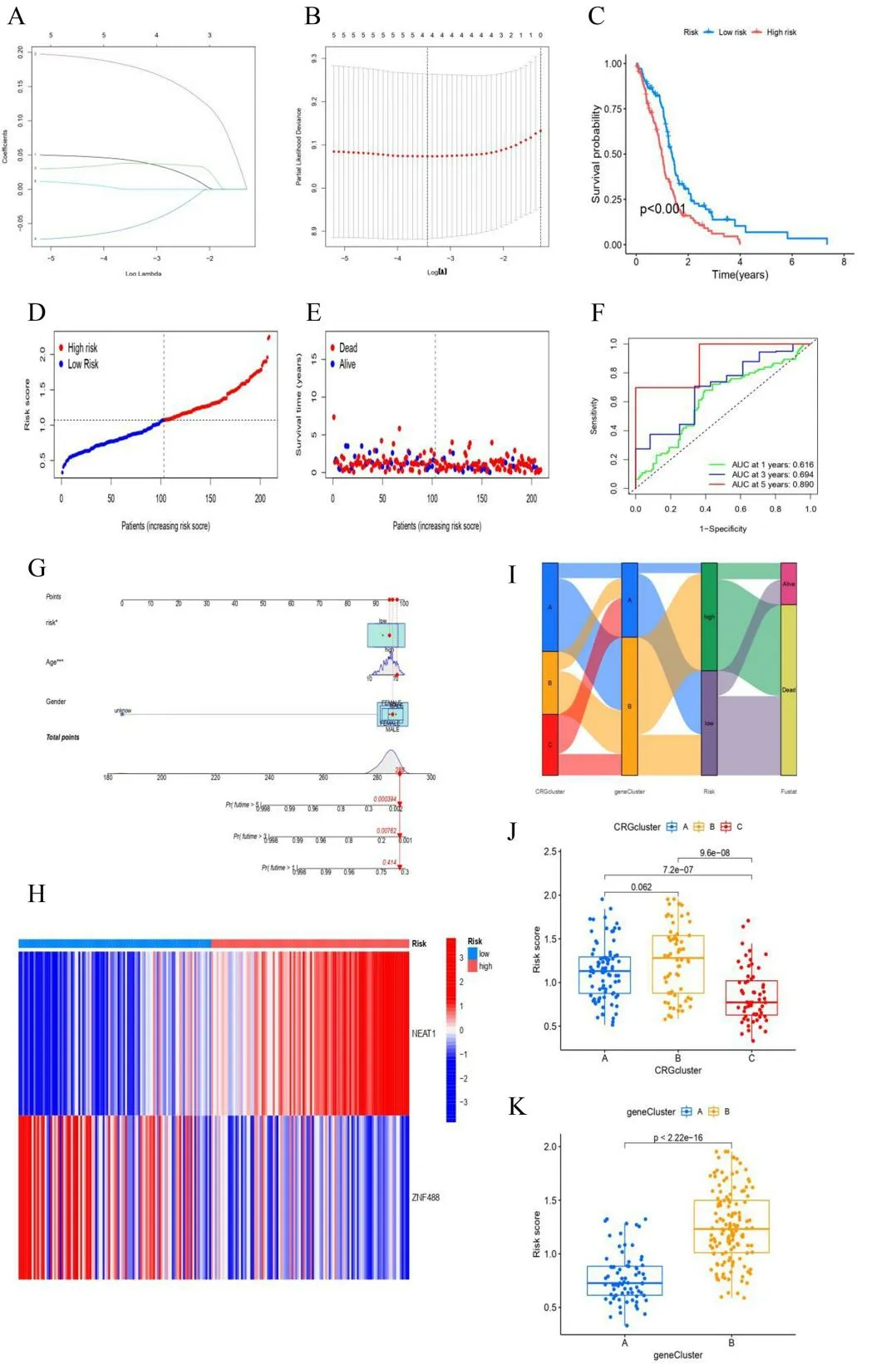
Figure 8. DEGs prognostic signature. (A, B) The process of constructing the prognostic signature. (C) The difference in survival probability between the high-risk group and low-risk group was shown via Kaplan-Meier curves(p <0.001),High-risk group was shown in red,low in blue.(D,E) The distributions of risk scores and OS status. Alive status was shown with blue dots,dead with red dots. (F) ROC curves showed the predictive efficiency of the risk signature (1-year AUC = 0.616,3-year AUC = 0.694,5-year AUC = 0.890). (G) A nomogram for predicting 1-,3-, and 5-year survival. (H) The heatmap of NEAT1 and ZNF488 expression levels in high-risk group and low-risk group. (I) Sankey diagram. (J, K) CRGclusters and geneclusters samples are scored using the coefficients.
Drug sensitivity analysis
To further investigate the sensitivity of drugs to patients in the high-risk and low-risk groups, a drug sensitivity analysis was performed. The results showed that ABT.888, AP.24534,and other 30 drugs with 50% inhibiting concentration (IC50) in the high-risk score group were lower than that in the low-risk score group, indicating higher drug sensitivity in the high-risk score group. IC50 of 11 drugs,such as AICAR and elesclomol,were lower in the low-risk score group than in the high-risk score group,indicating low drug sensitivity in the high-risk score group (Table 1).
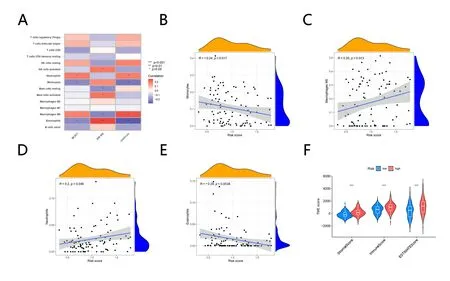
Figure 9. Correlation analysis of immune cells and differences in tumor microenvironment (A) Correlation analysis of immune cells. (B-E)Scatterplot of immune cells and risk. (F) Violin Plot shows TME score difference between high and low-risk groups.
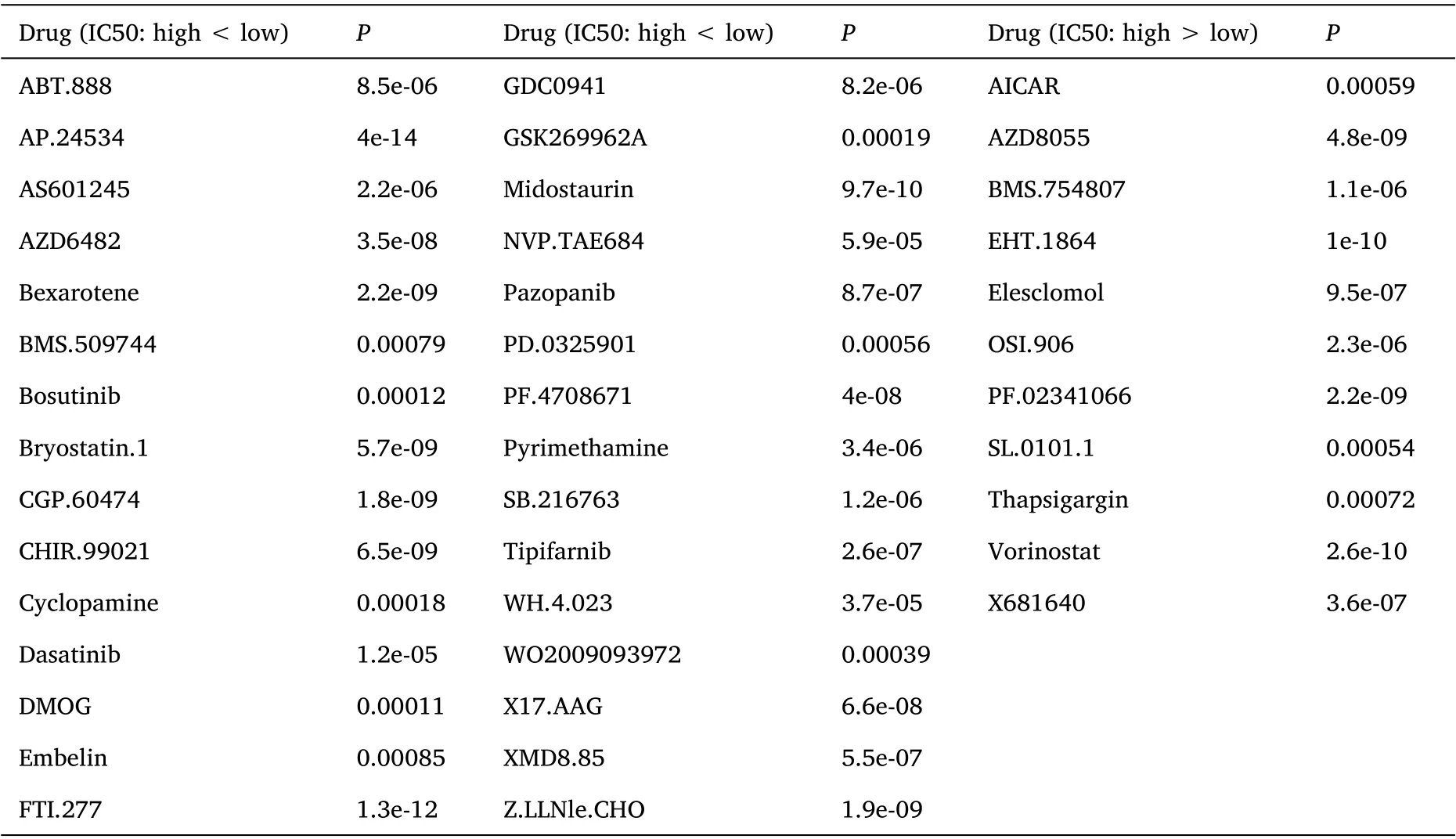
Table 1. Drug sensitivity analysis:41 drugs had different sensitivities in the high and low-risk score groups
Discussion
GBM is a serious threat to human physical and mental health due to high morbidity and mortality. Precision-targeted therapy and immunotherapy are one of the main research directions[13].In recent years, the role of cuproptosis in tumors has attracted much attention.Researchers have revealed the prognostic value of CRGs in renal clear cell carcinoma [14]. However, how CRGs regulate GBM remains unclear. Among the 19 CRGs, 7 genes, such as NFE2L2, were up-regulated in GBM, and 3 genes such as ATP7B were down-regulated in GBM.Erythroid 2 like 2(NFE2L2),a nuclear factor,is an antioxidant oxidase-related transcription factor that regulates oxidative stress and inflammation, and its high expression is a risk factor for many cancers, reducing OS [15]. The ATPase copper transporting beta (ATP7B) plays an important role in the normal physiological process of the human body and can remove excess copper from the body through the liver. Low expression of this gene can lead to severe nerve damage [16]. CNV results showed that CDKN2A and ATP7B were the genes with the highest copy number loss frequency. Studies have revealed that the absence of CDKN2A can promote the activation of CD4/6 and downstream transcription factor E2F1,then stimulate the upregulation of the BRN2 gene in melanoma,leading to tumor metastasis [17]. The high expressions of ATP7A,DLD, LIPT2 NFE2L2, and SLC31A1 can reduce GBM patients' survival rate. The high expressions of ATP7B, CDKN2A, DBT, DLST GCSH, and MTF1 can increase GBM patients'survival. CRGs are closely related to the prognosis of GBM.
Unsupervised cluster analysis was conducted according to the expression amount of CRGs in each sample, and all samples were divided into three clusters. Cluster B had the worst prognosis. A variety of immune cells accumulate in cluster B,such as activated CD8 T cells, immature dendritic cells, natural killer cells, etc. Activated CD8 T cell, also known as cytotoxic T cell (CTL), plays an important role in anti-tumor. On the one hand, it can produce cytokines such as TNF-α and IFN-γ. TNF-α can bind to TNFR indicated by target cells,activate the caspase pathway, and induce apoptosis of target cells.IFN-γ can increase antigen presentation by promoting the expression of MHC molecules on the surface of target cells, and can also improve the phagocytosis of macrophages and dendritic cells. On the other hand,CTL can lyse target cells by releasing perforin,grease,and other cytotoxic molecules [18]. Immature dendritic cells are derived from CD34+ hematopoietic podocytes. They absorb antigens by phagocytosis and endocytosis.Then mature into mature dendritic cells that are more conducive to presenting antigens. More efficient and strict technical methods of DC vaccine preparation and antigen load are continuously derived [19]. NK cells, derived from bone marrow lymphoid stem cells, are effector cells similar to CTL, which can inhibit tumor proliferation, migration, and other biological processes and play a cytotoxic role.In addition,NK cells produce a large number of cytokines, such as IFN-γ, which regulate adaptive immune processes [20]. Studies have revealed the potential of NK cells in adoptive immunotherapy of glioblastoma [21]. Drug metabolism cytochrome P450, ABC transporters, apoptosis high expression. were enriched by cluster B. Inhibition of cytochrome P450 reduced angiogenesis in glioblastoma multiforme rats [22]. ATP binding cassette (ABC) transporters regulate the transport of a variety of compounds through biofilms, consisting of 49 proteins. In the brain,they are expressed by microglial cells, astrocytes, neurons, and pericytes. Many chemotherapeutic agents are also substrates of ABC transporters that inhibit their entry into tumor cells [23]. High expression of the ABC transporters family, especially ABCA13, was found to be an independent prognostic factor in GBM patients [24].Apoptosis is to have a nuclear cell in apoptotic stimulation signal action through starting the mechanism of death inside the cell,through a series of signal transduction ways,and finally producing the process of cellular procedural denaturation and death. Apoptosis plays an important role in tumor genesis and development, and chemotherapy, radiotherapy, and biotherapy are mainly used to induce apoptosis of tumor cells. Scientists have also attempted to promote glioblastoma cell apoptosis in a variety of ways [25, 26].Twenty-one DEGs were selected from three clusters. For KEGG,ferroptosis, porphyrin metabolism, and legionellosis are the main ways enriched.For GO,oligodendrocyte development,central nervous system myelination, and axon ensheathment in the central nervous system are the main biological process. The differential genes in different clusters involve many different biological processes and pathways,which may be related to the generation of different clusters,but the specific mechanism remains to be further explored.
Based on two DEGs, we constructed a prognostic model. Two genes(NEAT1, and ZNF488) had independent predictive values in the prognostic model. The nuclear paraspeckle assembly transcript 1(NEAT1) is derived from a gene locus known as multiple endocrinomatosis types I on human chromosome 11, transcribed by RNA polymerase II,an uncut non-coding RNA with a length of about 4 kb [27]. NEAT1 is closely related to tumor cell cycle, apoptosis, and proliferation. Inhibition of NEAT1 expression induces G1 cell cycle arrest in non-small cell lung cancer [28]. The effect of promoting tumor cell proliferation has been demonstrated in pancreatic cancer[29]. Inhibition of NEAT1 expression and induction of G1 cell cycle arrest has also been demonstrated in gliomas [30]. The decrease of NEAT1 expression can block the proliferation of glioma stem cells(GSCs) by NEAT1/ LET-7G-5P /MAP3K1 axis and reduce the resistance to TMZ [31]. Glioma patients with high expression of NEAT1 tend to have larger tumor size, higher WHO grade, and tumor recurrence rate. Overexpression of NEAT1 is an independent prognostic factor for postoperative chemoradiotherapy and WHO grade of glioma and stage III-IV patients with high expression of NEAT1 and patients requiring postoperative chemoradiotherapy have a poorer OS [32]. Our study revealed that patients with high NEAT1 expression had a higher risk score,and NEAT1 may be a key indicator of the prognosis of GBM patients.These findings suggest that targeting NEAT1 may provide new therapeutic opportunities for patients with GBM. Zinc Finger protein 488 (ZNF488) is considered to be an oligodendrocyte-specific transcriptional inhibitor that plays an important role in myelin regeneration. The role of ZNF488 in nasopharyngeal and pancreatic cancer has been explored[33,34],but not in GBM. This study revealed that patients with low expression of ZNF488 had higher risk scores, and the relationship between ZNF488 and GBM may be one of the future research directions.
We conducted correlation analysis on NEAT1,ZNF488,and immune cells in the samples,then found that neutrophils and macrophages M0 were significantly positively correlated with a risk score, while monocytes and eosinophils were significantly negatively correlated.These immune cells may also be important signals for identifying high and low risk. The microenvironment of GBM is rich in a variety of immune cell types, including neutrophils and macrophages. A higher Neutrophil-lymphocyte ratio was an independent prognostic factor to a shorter overall survival of GBM [35]. Neutrophils were observed to promote the proliferation and migration of glioblastoma-initiating cells, leading to tumor progression [36]. Accumulating evidence indicates that macrophages promote glioma growth and invasion[37].Our study found that different risk scores have different immune landscapes, which may provide new ideas and targets for immunotherapy of GBM in the future. The analysis of the tumor microenvironment (TME) showed that the high content of stromal cells and immune cells, and the low purity of tumor samples in the high-risk group may be responsible for the poor prognosis. Drug sensitivity analysis showed that 41 drugs were different in the high and low-risk groups, which provided new ideas and new options for the treatment of GBM.
This study also has the following limitations: Firstly, access to transcriptome data and clinical data from open data sets will ignore intra-tumor heterogeneity.Secondly, this study revealed genes related to tumor prognosis,but the process of their regulation of GBM remains to be further studied. Thirdly, this study is a bioinformatics study of multiple groups of students, and all data are from public data sets,lacking in vivo and in vitro experimental verification. Future research will focus on this aspect.
Conclusions
By analyzing the expression of CRGs in GBM, this study identified three regulatory patterns of cuproptosis in GBM patients. DEGs of three clusters were obtained, and two key genes with independent prognostic values were found.Then we established prognostic models.A risk score can predict prognosis well, and it is also of certain significance in predicting drug sensitivity.
 Precision Medicine Research2022年3期
Precision Medicine Research2022年3期
- Precision Medicine Research的其它文章
- Prognostic implication and oncogenic role of RHCG in lung adenocarcinoma
- Correlation of bisphenol A action on genes interfering with estrogen signaling pathways and the development of endometriosis
- Identification of overlapping differentially expressed genes in hepatocellular carcinoma,breast cancer,and depression by bioinformatics analysis