
提取方法对汉麻叶大麻素成分及体外降糖降脂功能的影响*
2022-10-13 09:51李德海马可张道明尹静
中山大学学报(自然科学版)(中英文) 2022年5期
李德海,马可,张道明,尹静
1. 东北林业大学林学院,黑龙 江哈尔滨 150040
2. 黑龙江省森林食品资源利用重点实验室,黑龙 江哈尔滨 150040
汉麻Cannabis sativaL.是一种一年生雌雄异株的草本植物,早期汉麻因其茎秆、叶子中含有丰富的纤维被广泛用于纺织工业中。近年来汉麻种子、叶子和其副产物中含有丰富的蛋白质、油脂、大麻素和非大麻素等成分而在化妆品、药品和食品工业中得到广泛研究与应用。虽然汉麻因含有致幻成瘾的四氢大麻酚(THC)导致在许多地区被禁止使用,但THC 在医药治疗疾病时可以用作麻醉剂[1]。为避免非法使用大麻作为毒品来娱乐吸食等,仅允许培养wTHC< 0.2%的工业大麻[2]。研究表明汉麻中含有丰富的大麻二酚(CBD)、大麻色烯、大麻环萜酚等大麻素类化合物,具有防治慢性疾病、增强记忆力、改善睡眠、降血糖血脂与抗氧化等作用。其中CBD 在高血糖状态下对人体视网膜微血管内皮细胞(HRCECs)的增长具有一定的抑制作用,进而影响核转录因子(FOXO3a)的活性,最终抑制血管内皮生长因子(VEGF)的表达[3]。内源性大麻素系统可以降低肥胖小鼠空腹血糖、空腹胰岛素水平,通过拮抗剂抑制大麻素受体2(CB2 受体)起到控制肥胖和调节血糖水平的作用[4]。另外大麻籽油中含有大麻二酚、大麻酚、大麻萜酚等主要成分,可以显著降低血清和肝脏的Lee′s 指数、总胆固醇、甘油三酯、低密度脂蛋白水平、丙二醛含量、肾脏和附睾周围脂肪组织的含量,提高血清和肝脏的高密度脂蛋白水平,增强超氧化物歧化酶活性,具有显著的降血脂功效[5]。可见大麻酚类化合物具有调节血糖血脂的功能,对高血脂高血糖患者有辅助调节作用。
大麻素是大麻植株特有的活性成分,其中wCBD仅有1%~3%,这部分物质的提取、分离纯化是该研究领域的难题,选择适用于大麻素的提取方法对其纯度的提高与产品开发利用具有深远影响。目前关于火麻仁油中提取大麻素类化合物的报道指出,利用超声波辅助提取法确定最佳提取工艺,使火麻仁油中大麻素类化合物提取得较完全,为其中大麻酚类化合物的含量测定奠定基础[6]。以火麻叶为原料采用热回流法提取大麻素中的大麻二酚,此法对浸膏得率与wCBD有显著影响,但因需要加热,故有消耗能量且费时费力的缺点[7]。为了进一步提高大麻素的提取效率,通过对超临界二氧化碳萃取温度、萃取时间、夹带剂用量、萃取次数进行筛选,提高了3 种大麻酚的提取率,并且具有优良的抑菌性能[8]。但是超临界二氧化碳设备投资大、能耗高,显著提高了制备成本。近年来,生物酶法、超声波[9]与高剪切辅助[10]等提取方式在成分提取方面越来越受到大家的关注,通过细胞壁分解酶对工业大麻叶进行酶解,经过溶剂萃取、脱色等过程,能够大大提高大麻二酚的提取率[11]。采用超声波技术从汉麻花中提取生物活性化合物多酚、黄酮与大麻素,使抗氧化活性有明显的增强[12]。高剪切辅助提取法[13]至今并未有研究用于汉麻提取,因易操作且设备简单、提取效率高、节约能源等优点,越来越受到重视。
因此本试验为建立一种高效且保持大麻素活性的制备方法,以小叶汉麻为原料,探究了溶剂提取法、酶辅助提取法、超声波辅助提取法、高剪切辅助提取法4 种提取方式对大麻素浸膏得率、wCBD以及体外降血脂血糖的影响,旨在为推动汉麻资源的利用及开发具有调节血糖血脂作用的健康产品提供科学依据。
1 材料与方法
1.1 材料与试剂
小叶汉麻:产自黑龙江青冈县,取叶子进行60 ℃烘干,粉碎,过60目筛备用。
试剂:对硝基苯-α-D-葡萄糖苷(PNPG),上海麦克林生化科技有限公司;牛磺胆酸钠、甘氨胆酸钠、胆酸钠,上海金穗生物科技有限公司;α-淀粉酶(76 000 U/mg)、α-葡萄糖苷酶(70 万U/mL)、纤维素酶(50 U/mg)、果胶酶(500 U/mg)、胃蛋白酶(3 000 U/mg)、胰蛋白酶(75 U/mg),上海源叶生物科技有限公司;大麻二酚标准品,云南汉素生物科技有限公司;四氢大麻酚标准品,美国Sigma公司;甲醇为色谱级;其他试剂均为国产分析纯。
1.2 仪器与设备
Thermo Fisher Q Exactive Focus 型超高效液相质谱联用仪,美国Thermo公司;Agilent 1260型高效液相色谱仪,美国Agilent 公司;TGL-16G 台式离心机,上海安亭科学仪器厂;AL-1041C 分析天平,瑞士Mettler Toledo 公司;DK-8D 电热恒温水槽,上海森信实验仪器有限责任公司;RE-52旋转蒸发器,上海亚荣生化仪器公司;KQ-500DE 型数控超声波清洗器,昆山市超声仪器有限公司;FLUKO 高剪切乳化机,上海弗鲁克流体机械制造有限公司;EPOCH12 型酶标仪,美国伯腾仪器有限公司。
1.3 试验方法
1.3.1 不同提取方法制备汉麻叶大麻素浸膏 取一定量汉麻叶粉,分别采用以下4 种方法进行处理。
溶剂提取法[14]按照料(m,g)∶液(V,mL)=1∶4分别加入提取剂(φ=70%乙醇、φ=80%乙醇、φ=90%乙醇、φ=95%乙醇、无水乙醇、石油醚、正己烷,以及V乙醇∶V二氯甲烷=4∶6),在25 ℃下转速为500 r/min的搅拌器中搅拌2.0 h。
酶辅助提取法根据参考文献[15]稍做修改,预实验基础上,使用pH 5.0 的磷酸盐缓冲溶液配制酶液。按料(m,g)∶液(V,mL)=1∶10 加入酶液,酶的添加量分别为:纤维素酶250 U/g(汉麻叶粉)、果胶酶2 500 U/g、纤维素果胶复合酶2∶1(500 U/g∶2 500 U/g)。在温度50 ℃摇床内分别酶解时间(0.25、0.5、0.75、1.0 h),酶解后放入100 ℃中灭活5 min。取出冷却至室温,4 000 r/min 离心15 min 取沉淀,再加入料(m,g)∶液(V,mL)=1∶4的最佳提取溶剂(由溶剂提取法确定),然后将其置于转速为500 r/min的搅拌器中快速搅拌3 h。
超声波辅助提取法[16]以料(m,g)∶液(V,mL)=1∶4 加入最佳提取溶剂(由溶剂提取法确定),超声温度30 ℃,考察超声时间(20、25、30、35、40 min)、超声功率(180、240、300 W),超声处理后溶剂提取30 min。
高剪切辅助提取法[17]以料(m,g)∶液(V,mL)=1∶4 加入最佳提取溶剂(由溶剂提取法确定),高剪切速率分别为(12 000、14 000、16 000、18 000 r/min),剪切时间(30、60、90、120、130 s)启动高剪切10 s,暂停20 s为1个周期,溶剂再提取30 min。
以上所有样品处理后,全部过滤取滤液。平行试验3 次,得提取液,在60 ℃、0.05 MPa 条件下进行旋蒸除掉溶剂,得到膏状物,该物质即为含有汉麻素的浸膏。分别以大麻素浸膏得率、wCBD、wTHC为指标来评价提取效果。
1.3.2 汉麻叶中大麻素浸膏得率测定 分别称取汉麻叶粉,按1.3.1方法,在设定的温度、时间等条件下提取,冷却抽滤,将滤液在60 ℃下旋蒸浓缩制得浸膏。浸膏得率为
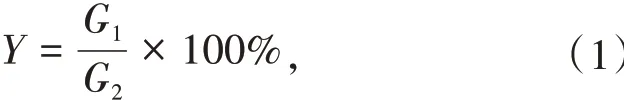
式中Y为浸膏得率(%);G1为浸膏的质量(g);G2为汉麻叶粉的质量(g)。
1.3.3 汉麻浸膏中CBD 与THC 含量测定 采用高效液相色谱法检测,取汉麻叶的大麻素浸膏用甲醇配制成质量浓度1 mg/mL,微孔滤膜滤过,取滤液作为供试品溶液。参考孙孔春等[18]的方法,稍做修改。标准曲线的绘制:称取10.000 1 mg 大麻二酚标准品,以甲醇定容制成母液(ρ=1 mg/mL),分别取20、40、80、120、160、200 μL,均放置于2 mL 容量瓶中,配制成ρ为0.01、0.02、0.04、0.06、0.08、0.1 mg/mL的标准品溶液。按条件进行检测,以大麻二酚质量浓度X为横坐标,峰面积Y为纵坐标绘制标准曲线。Y=11.87X-40.126,R2=0.990 6,同以上方法配置四氢大麻酚标准品,绘制标准曲线Y=22.172X-1.536,R2=0.999 9。适用范围:0.01~0.1 mg/mL。色谱条件:色谱柱,Kromasil C18柱(4.6 mm×250 mm,5 μm);流速,1.0 mL/min;检测波长,230 nm;进样量,10 μL;柱温,30 ℃;以V甲醇∶V水=85∶15 为流动相进行等度洗脱。在此色谱条件下,取大麻素样品进样分析。
1.3.4 超高效液相质谱联用技术对浸膏中大麻素类成分分析 取4 种提取方法制备的汉麻叶浸膏0.001 0 g 用甲醇定容至10 mL,8 000 r/min 离心20 min,0.22 μm 微孔滤膜过滤,取滤液作为样品溶液备用。UPLC-Q Exactive Focus MS/MS 分析条件:使用GOLD VANQUISH 色谱柱,100 mm×2.1 mm,1.9 μm;流动相A 为甲酸水溶液,φ=0.1%,B 为甲醇溶液;梯度洗脱的设置如下:从φ=2% 到φ=98%B 的线性梯度为0.0~9.0 min;9.1~12.0 minφ=98%B;12.1~15.0 min 回到φ=2%B,平衡柱5 min,总运行时间为15 min。柱温40 ℃;流速设定为0.350 mL/min;样品进样量为5µL。
质谱同时在正、负离子模式下检测:电喷雾离子化电离源(HESI);毛细管温度320 ℃;汽化器温度350 ℃;电喷雾电压3 kV;鞘气体积流量40 个单位;辅助气体积流量10 个单位;质谱分辨率17 500 FWHM;扫描范围为m/z80~1 200;AGC设置为1e5;NCE值分别为20、40和60 eV。
采用Compound Discoverer 3.2软件和compounds化学成分数据库信息进行匹配,初步进行分析,与ChemSpider、mzCloud、mzVault 等数据库对比。通过质谱图、相对分子质量、一级、二级碎片离子和化学结构与参考文献信息分析后确定物质名称与分类[19]。
1.3.5 大麻素类体外降血糖功能的研究α-葡萄糖苷酶抑制率的测定:取50 μL 0.5 U/mL 的α-葡萄糖苷酶溶液置于96 孔板中,再分别加入不同质量浓度(0.05、0.1、0.2、0.4、0.8、2.0、5.0 mg/mL)的大麻素样品液100 μL,于37 ℃水浴10 min,加入10 mmol/L PNPG 50 μL,再放入37 ℃水浴锅中加热10 min,最后使用0.1 mol/L Na2CO3100 μL 终止反应,在酶标仪上于405 nm 处测定吸光度A1;另取100 μL pH 6.8 磷酸盐缓冲液代替大麻素样品液测定吸光度A2;为消除反应体系干扰,在不加α-葡萄糖苷酶溶液条件下分别测定只加入大麻素样品液反应体系的吸光度A3和只加入磷酸盐缓冲溶液反应体系的吸光度A4。IC50值是通过不同质量浓度大麻素样品液对α-葡萄糖苷酶抑制率的计算而得。
α-淀粉酶抑制率的测定:取50 μL 0.5 U/mL的α-淀粉酶液溶液置于96 孔板中,再分别加入不同质量浓度(0.05、0.1、0.2、0.4、0.8、2.0、5.0 mg/mL)的大麻素样品液100 μL,放入37 ℃水浴锅中10 min,加入w=0.2%淀粉溶液50 μL,再次37 ℃水浴10 min 后,使用显色剂(5 mmol/L I2和5 mmol/L KI 溶于1 mol/L HCl)100 μL 终止反应,在酶标仪上于620 nm 处测定吸光度A1;另取100 μL pH 6.8 磷酸盐缓冲液代替大麻素样品液,测定吸光度A2;再测定只有大麻素样品液反应体系的吸光度A3和只有磷酸盐缓冲溶液反应体系的吸光度A4。IC50值是通过不同质量浓度大麻素样品液对α-淀粉酶抑制率的计算而得。
每组试验3次平行,公式如下

式中A1为样品组吸光度,A2为样品空白组吸光度,A3为样品对照组吸光度,A4为空白对照组吸光度。
1.3.6 大麻素类体外降血脂功能的研究 胆酸盐结合能力的测定:准确吸取1 mL 大麻素样品液于试管中,依次加入1 mL 10 mg/mL 胃蛋白酶(以pH 6.3 的0.1 mol/L 磷酸缓冲液配制),3 mL 0.01 mol/L的HCl溶液,模拟胃环境,在37 ℃恒温振荡消化1 h;以0.1 mol/L 的NaOH 溶液调节pH 值至6.3,随后加入4 mL 10 mg/mL 胰蛋白酶,模拟肠道环境,在37 ℃恒温振荡消化1 h,加入4 mL 1 mmol/L 胆酸盐(胆酸钠、牛磺胆酸钠、甘氨胆酸钠)消化1 h,8 000 r/min 离心20 min,取上清液,加入6 mLφ=60%的H2SO4于70 ℃水浴20 min,取出冰浴5 min 后,在387 nm 处测定吸光度Ae,另取1 mL pH 6.3的0.1 mol/L磷酸缓冲液代替大麻素样品液,测定吸光度A0;再测定只有大麻素样品液反应体系并用磷酸缓冲液代替H2SO4的吸光度Ai。IC50值是通过不同质量浓度大麻素样品液对胆酸盐结合率的计算而得,平行3次实验,计算样品对胆酸盐结合率[20]计算公式如下

式中A0为样品空白组吸光度,Ae为样品组吸光度,Ai为样品对照组吸光度。
1.4 数据处理与分析
每个处理进行平行3次试验,结果表示为平均值±标准差。采用Origin、SPSS 软件对数据进行处理与分析,利用置信度为95%的最小显著性差异(P<0.05)比较平均值。
2 结果与讨论
2.1 提取方法对大麻素浸膏得率影响
图1(a)~(d)通过汉麻浸膏得率、wCBD、wTHC为评价指标,得到4种提取方法的最佳提取条件。分析图1(a)可知,大麻素浸膏得率随着乙醇体积分数的增加而增大,其中无水乙醇制备的大麻素浸膏得率最大,为9.43%,其次是φ=95%乙醇制备的浸膏得率为3.95%。而乙醇/二氯甲烷制备的浸膏得率为3.25%,显著低于无水乙醇和φ=95%乙醇分别制备浸膏的得率,正己烷和石油醚制备的浸膏得率效果更低。汉麻浸膏中wCBD检测结果表明φ=95% 乙醇制备的浸膏中wCBD最高,值为241.80 mg/g,显著高于无水乙醇制备的汉麻浸膏wCBD。无水乙醇作为提取溶剂会导致汉麻叶中更多非大麻素类化合物溶出,导致汉麻浸膏量增加,而浸膏中wCBD降低会影响大麻素的纯度,不利于后续大麻素类化合物的分离纯化和浓缩单元操作,增大工作量和能耗[21],因此本实验选取φ=95%乙醇为最佳提取剂。由图1(b)可知,浸膏得率随着超声时间的增加呈现先升高后降低的趋势,当超声功率为300 W 时,超声时间30 min 时浸膏得率达到最大,为10.03%。其次是超声功率为240 W时,超声时间为25 min 浸膏得率较大为8.68%。当超声功率为180 W 时,超声时间为30 min 浸膏得率为7.75%,显著低于前两种条件浸膏得率。当超声功率为300 W,超声时间25 min时浸膏得率7.06%,但是大麻素浸膏中wCBD检测值为283.04 mg/g,显著高于超声功率为300 W,超声时间30 min 时大麻素浸膏wCBD。随着超声功率的增大,细胞的破碎程度变大,但是过大的超声功率会使大麻素类化合物被破坏,过多杂质溶出。适宜的超声时间会使细胞中的物质更好地溶出,如果时间过长或过短都会对大麻素有影响[22-23]。经过各方面考虑,选用超声功率300 W、超声时间25 min作为超声提取最佳条件。
图1(c)实验结果表明,随着酶解时间延长得率逐渐增大,直到1 h 达到最大,随后逐渐降低。经过纤维素酶和果胶酶处理的样品,酶解1 h 其浸膏得率最大分别为19.90%、19.98%,这是因为纤维素酶和果胶酶破坏了植物细胞壁组织,从而让溶剂更好地渗入组织内部[24],进而提高大麻素的提取。并且这两种酶的效果差异性不显著,这表明汉麻叶细胞壁主要成分是纤维素和果胶。纤维素与果胶进行复合酶解1 h,其浸膏得率达到了21.11%,浸膏中wCBD检测值最高为w=24.98 mg/g,其效果明显优于单一酶,选用复合酶酶解时间1 h作为酶辅助提取法提取最佳条件。如图1(d)所示,在4个速率条件下,浸膏得率随时间的增加而先升高后降低。高剪切速率为16 000 r/min 时,高剪切时间90 s 时,浸膏得率达到最大值7.5%,其次高剪切速率为14 000 r/min 时间120 s 时浸膏得率为7.25%,而高剪切速率为14 000 r/min 时间130 s 时浸膏得率为6.93%,这是由于相同速率条件下高剪切时间过长对目标物具有破坏性。高剪切速率14 000 r/min、高剪切时间60 s 时浸膏得率为3.00%,显著低于其他条件下浸膏得率,但汉麻浸膏中wCBD检测值最大为161.46 mg/g。在设定的剪切频率范围内,随着剪切频率增大,机械剪切效应增大,气蚀作用增强,导致非大麻素类物质的溶出[25]。因此,选用高剪切速率14 000 r/min、高剪切时间60 s作为高剪切提取最佳条件。

图1 不同提取方法对浸膏得率的影响Fig.1 Effects of different extraction methods on the yield of extract
2.2 HPLC 法测定不同提取方法样品中的CBD 和THC的含量
4 种提取方法的汉麻中大麻素含量结果见表1所示,通过4种样品高效液相色谱图进行比对。由表1可知,对于大麻素,超声波辅助提取法可以得到最高含量,且与其他3 种方法有显著差异(P<0.05),其中超声波辅助提取法wCBD最大,其值为(283.04±16.63)mg/g,其次是溶剂提取法与高剪切辅助提取法,而酶辅助提取法wCBD最低,其值仅为(24.98±3.24)mg/g。超声技术由于具有机械效应、空化效应和热效应,通过增大分子的运动速度与穿透力来增加提取效果[26]。而高剪切技术由于转子高速旋转产生强烈的剪切力和离心挤压力等机械作用,使汉麻叶组织细胞发生变形,细胞壁破裂,加速细胞内物质溶出,虽提高了提取效果[27],但是也对大麻素造成部分破坏。采用溶剂提取法直接提取,获得wCBD也比较好,但耗费时间长,不能够快速提取大麻素。通过上述4种方法处理后发现,易使人致幻成瘾的THC 完全被去除,为以后汉麻叶的食品、药品等应用提供科学的依据。
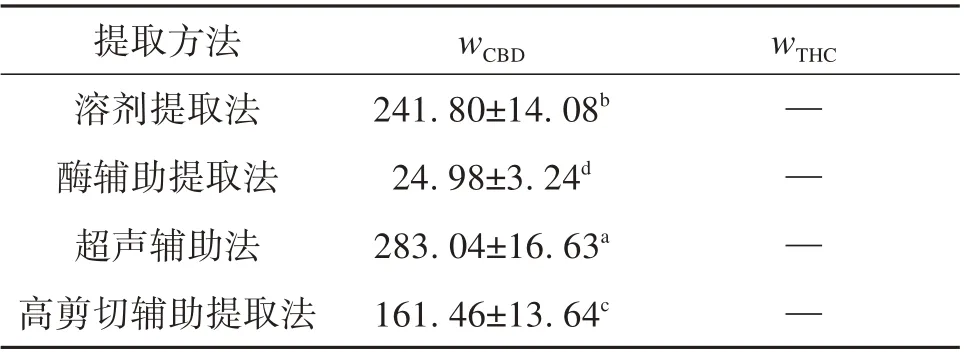
表1 不同提取方法下浸膏中wCBD和wTHC测定结果1)Table 1 Determination results of CBD and THC content in extract with different extraction methods mg/g
2.3 液相-质谱联用对提取方法萃取物成分分析
2.3.1 UPLC-Q Exactive Focus MS/MS 分析结果采用UPLC-Q Exactive Focus MS/MS 超高效液相高分辨质谱联用仪对汉麻叶提取物进行成分分析,所得到的总离子流质谱图如图2。按照上述数据处理方法对汉麻叶中的大麻素类化合物进行结构推测和确证。目标物存在较多的同分异构体,主要通过质谱裂解方式的不同进行区分。依据化合物的高分辨质谱数据,结合自建数据库等多个数据库比对,12 种物质结合质子分子,总结了准确的相对分子质量、保留时间、分子式和二级碎片,具体鉴定结果见表2。
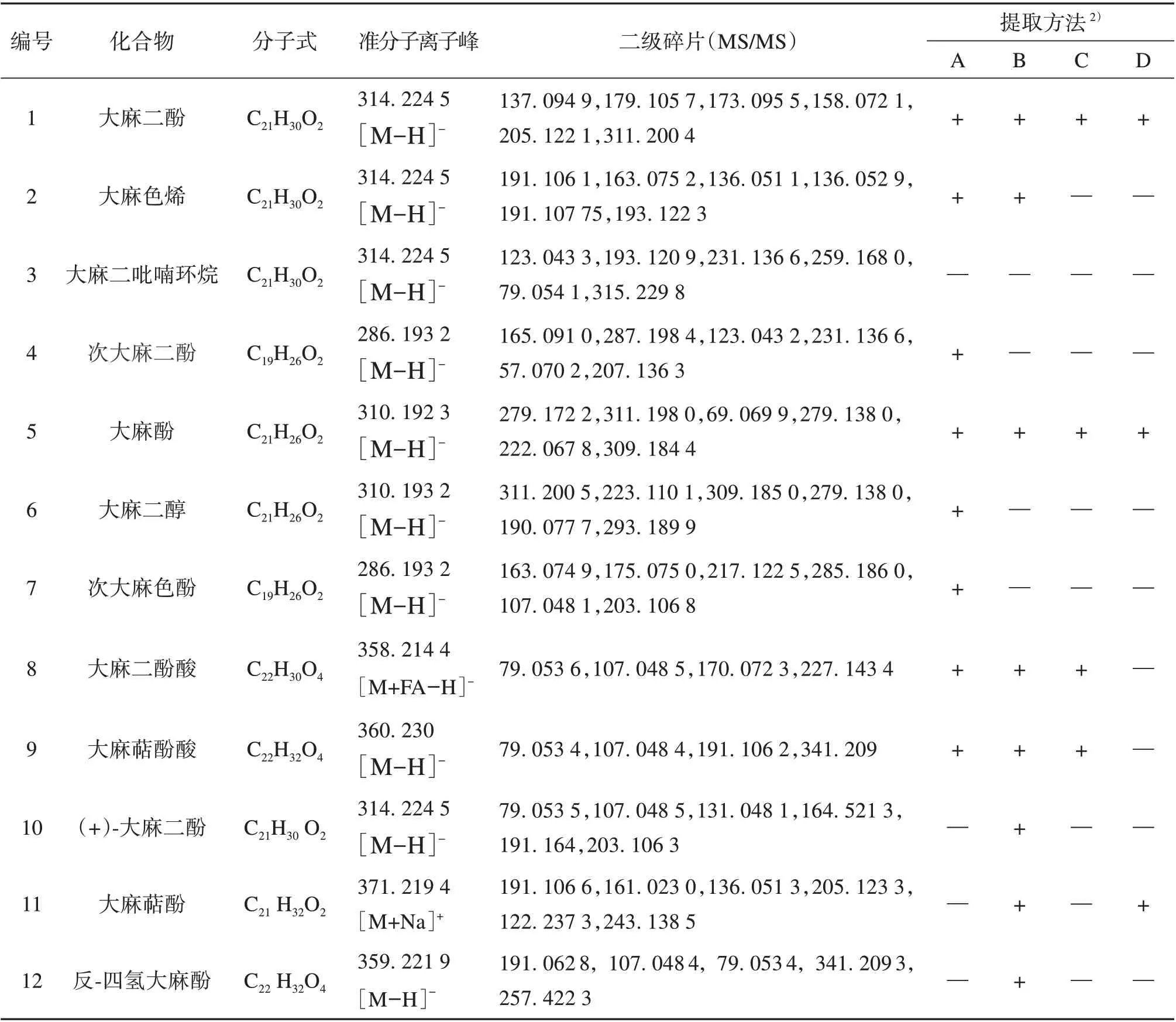
表2 汉麻叶大麻素类成分鉴定1)Table 2 Identification of cannabinoids in hemp leaves

图2 汉麻叶4种方法提取物总离子流质谱图(TIC)Fig. 2 Total ion mass spectrometry of hemp leaves extracted by four methods(TIC)
2.3.2 大麻素类化合物成分鉴定 研究共分析鉴定了12 种大麻素类化合物,以含量较多的大麻二酚和大麻色烯为例,二者互为同分异构体。大麻二酚MS1谱图中显示该化合物的准分子离子峰为m/z314.224 5[M-H]-,可以作为大麻素类化合物的特征离子,特征离子进一步失去1个H 形成,其分子式C21H30O2,相对分子质量为314.224 1,能够很好地吻合。MS2图谱给出主要的碎片离子分别为m/z137.094 9,179.105 7,173.095 5,158.072 1,205.122 1,311.200 4。大麻色烯MS1谱图中显示该化合物的准分子离子峰为m/z314.224 5[M-H]-,可以作为大麻素类化合物的特征离子,特征离子进一步失去1 个H 形成,其分子式C21H30O2,相对分子质量为314.224 1,能够很好地吻合。MS2图谱给出主要的碎片离子分别为m/z191.106 1,163.075 2,136.051 1,136.052 9,191.107 7,193.122 3。利用液相色谱质谱联用技术,Gul等[28]对大麻根中的10种大麻素类化合物,包括大麻色烯、Δ8-四氢大麻酚、大麻酚、大麻二酚等,进行了定量分析。Christinat 等[29]采用气相色谱质谱联用法,对大麻类食品中15 种大麻素类成分的含量进行了测定,包括大麻二酚、大麻萜酚、Δ9-四氢大麻酚等。
2.4 不同提取方法对大麻素的体外降血糖影响
α-葡萄糖苷酶和α-淀粉酶在糖类消化以及吸收中发挥着重要的作用,可抑制酶的活性、降低机体的血糖和血脂水平、防止餐后高血糖的发生,对2型糖尿病的预防及治疗具有积极作用[30]。
不同提取方法获得的大麻素浸膏对α-葡萄糖苷酶和α-淀粉酶抑制能力如图3 所示,4 种提取方法都呈现随样品浓度增大,抑制能力逐渐上升的趋势。浸膏质量浓度为0~2 mg/mL 时,4 种提取方法的抑制率快速增加,2 ~5 mg/mL 时抑制率增长缓慢。如图3(a)所示,在质量浓度为5 mg/mL 时,溶剂提取法对α-葡萄糖苷酶抑制率最强,为92.85%,其次是酶辅助提取法、超声波辅助提取法和高剪切辅助提取法, 分别为85.38%、77.78%、72.22%。溶剂提取法对原料无机械作用与高速剪切破坏,并且没有酶作用产生大量杂质,因此对α-葡萄糖苷酶抑制能力最好,对汉麻叶分子结构破坏程度较小。如图3(c)所示,通过软件计算α-葡萄糖苷酶的半抑制质量浓度分别为酶辅助提取法1.39 mg/mL、高剪切辅助提取法1.37 mg/mL、超声波辅助提取法0.20 mg/mL、溶剂提取法0.19 mg/mL,超声波辅助提取法与溶剂提取法之间差异不显著(P>0.05),超声波辅助提取法与酶辅助提取法和高剪切辅助提取法之间差异显著(P<0.05),酶辅助提取法与高剪切辅助提取法之间差异不显著(P>0.05)。分析图3(b)实验结果可知,当浸膏质量浓度为2 mg/mL 时,4 种提取方法制备大麻素浸膏对淀粉酶的抑制能力不同,其中高剪切辅助提取法抑制率为77.48%,溶剂提取法为71.51%,其次是超声波辅助提取法64.58%,酶辅助提取法抑制率最小37.57%。当浸膏质量浓度为5 mg/mL时,其对α-淀粉酶的抑制能力与质量浓度为2 mg/mL时抑制能力相同,高剪切辅助提取法具有最大的α-淀粉酶抑制能力,为88.22%,酶辅助提取法具有最小的抑制率为56.32%。不同提取方法的大麻素降血糖能力存在差异,这可能与大麻素类化合物中其他成分相关。见图3(c)可知,4 种提取方法的大麻素浸膏对α-淀粉酶抑制率IC50值由大到小为:酶辅助提取法为3.44 mg/mL、超声波辅助提取法为0.50 mg/mL、高剪切辅助提取法为0.21 mg/mL、溶剂提取法为0.13 mg/mL,高剪切辅助提取法与溶剂提取法之间的差异不显著(P>0.05),而酶辅助提取法与超声波辅助提取法之间存在显著性差异(P<0.05)。

图3 不同提取方法对大麻素的体外降血糖影响Fig. 3 Effects of different extraction methods on the hypoglycemic effect of cannabinoids in vitro
2.5 不同提取方法对大麻素的体外降血脂影响
分别采用溶剂提取法、酶辅助提取法、超声波辅助与高剪切辅助提取法制备大麻素,按照方法1.3.6,吸附结合胆酸盐,并比较分析4 种不同提取方法获得大麻素结合甘氨胆酸钠、胆酸钠、牛磺胆酸钠的差异。
由图4 可见,4 种方法提取的大麻素结合3 种胆酸盐能力其结合量和质量浓度之间呈明显的剂量效应关系,随着质量浓度的增加其结合量逐渐上升。由图4(a)所示,在浸膏质量浓度为15 mg/mL 时,除超声波辅助提取法其他3 种提取方法结合率变化较小,溶剂提取法与甘氨胆酸钠结合率最大为82.41%。当浸膏质量浓度为20 mg/mL 时,超声提取技术获得的大麻素能力最强为87.09%,降血脂功能最显著,其次溶剂提取法抑制率为83.18%、高剪切辅助提取法抑制率为67.82%、酶辅助提取法抑制率为36.64%。说明超声技术能促进大麻素的溶出,增强与甘氨胆酸钠的结合,加快甘氨胆酸钠在肠肝中的循环,胆酸盐排出时会促使肝脏不断把体内胆固醇转化成胆酸盐,促进胆固醇降解代谢,从而起到降血脂的作用[31]。如图4(d)所示,当浸膏质量浓度达到20 mg/mL,IC50值由大到小分别为酶辅助提取法24.41 mg/mL、高剪切辅助提取法9.31 mg/mL、超声波辅助提取法8.64 mg/mL、溶剂提取法6.52 mg/mL,酶辅助提取法与溶剂提取法之间存在显著性差异(P<0.05),高剪切辅助提取法与超声波辅助提取法之间差异不显著(P>0.05)。由图4(b)可见,在15 mg/mL 继续增加样品质量浓度,结合率下降,可能是由于浸膏质量浓度过高时,会抑制自身降血脂活性。其中超声波辅助提取法提取的大麻素对胆酸钠的结合率最大为88.85%,超声功率逐渐提高,对细胞壁的破损程度逐渐增大,使大麻素快速溶出,提取率升高,增大与胆酸钠的结合量,降血脂作用有一定的提升效果[32]。其次溶剂提取法胆酸钠结合率较大,为86.64%,高剪切辅助提取法为62.79%,酶辅助提取法胆酸钠结合率最小,为24.32%。如图4(d)所示,当浸膏质量浓度达到20 mg/mL,大麻素与胆酸钠结合率IC50值由大到小分别为酶辅助提取法56.58 mg/mL、高剪切辅助提取法11.01 mg/mL、溶剂提取法7.11 mg/mL、超声波辅助提取法4.88 mg/mL,各组间差异性显著(P<0.05)。由图4(c)可见,当浸膏质量浓度为20 mg/mL 时,4 种方法结合率达到最大。高剪切辅助提取法明显优于其他几种提取技术,其对牛磺胆酸钠的结合率为94.48%,主要是因为牛磺胆酸钠中磺酸基的酸性比胆酸钠和甘氨胆酸钠的羧基更强,在高浓度下更容易离子化,且由于高剪切技术使细胞被迅速破碎,大大减小了有效成分的扩散阻力,大麻素被快速释放,结合量明显增强,降血脂效果明显[33]。其次是溶剂提取法与超声波辅助提取法对牛磺胆酸钠的结合率分别为93.63%、73.75%,而酶辅助提取法与牛磺胆酸钠结合率最低为22.17%,汉麻叶细胞壁被酶解后使大量杂质溶出,破坏大麻素成分,大大降低提取效率,与牛磺胆酸钠的结合量明显降低,降血脂功能明显低于其他3种方法。如图4(d)所示,当浸膏质量浓度达到最大,牛磺胆酸钠结合率IC50值由大到小分别为酶辅助提取法32.91 mg/mL、超声波辅助提取法12.14 mg/mL、溶剂提取法4.83 mg/mL、高剪切辅助提取法3.26 mg/mL,各组间差异性显著(P<0.05)。

图4 不同提取方法对大麻素的体外降血脂影响Fig. 4 Effect of different extraction methods on the hypolipemia of cannabinoids in vitro
3 结 论
本文通过溶剂提取法、酶辅助提取法、超声波辅助与高剪切辅助4种提取方法制备大麻素类成分,结果表明超声波辅助提取法wCBD最高,为283.04 mg/g;其次是溶剂提取法,为241.80 mg/g;高剪切辅助提取法,为161.46 mg/g;酶辅助提取法最低,为24.98 mg/g。利用UPLC-Q Exactive Focus MS/MS分析可知超声波辅助提取法得到的大麻素成分最多,达到8种。体外降血糖、降血脂试验发现,超声波辅助提取法提取出的大麻素类成分对α-葡萄糖苷酶与α-淀粉酶都具有较好的抑制能力,且对胆酸盐结合能力最高。4 种方法对α-葡萄糖苷酶和α-淀粉酶的最好抑制率分别为92.85%和88.22%;不同方法对甘氨胆酸钠、胆酸钠和牛磺胆酸钠的结合率分别为87.09%、88.85% 和94.48%。研究对4 种提取方法进行比较发现,超声波辅助提取法获得的大麻素含量最高,并且都具有较为良好且稳定的降糖降脂功能。因此低成本、高效率的超声波辅助提取法在汉麻资源开发与应用中具有潜在优势。
猜你喜欢
健康体检与管理(2022年4期)2022-05-13
昆明医科大学学报(2022年3期)2022-04-19
医学前沿(2021年18期)2021-04-14
昆明医科大学学报(2021年2期)2021-03-29
现代食品·下(2020年9期)2020-11-06
商情(2020年32期)2020-07-23
家庭百事通·健康一点通(2018年9期)2018-10-12
上海医药(2017年13期)2017-07-20
儿童故事画报·发现号趣味百科(2017年1期)2017-06-01
华声(2016年20期)2016-11-19
