
SSR markers development and their application in genetic diversity evaluation of garlic (Allium sativum) germplasm
2022-10-12 06:48:38XiaxiaLiLijunQiaoBirongChenYujieZhengChengchenZhiSiyuZhangYupengPanZhihuiCheng
植物多样性 2022年5期
Xiaxia Li,Lijun Qiao,Birong Chen,Yujie Zheng,Chengchen Zhi,Siyu Zhang,Yupeng Pan,Zhihui Cheng
College of Horticulture, Northwest A&F University, Yangling, 712100, Shaanxi Province, China
A B S T R A C T Garlic (Allium sativum), an asexually propagated vegetable and medicinal crop, has abundant genetic variation. Genetic diversity evaluation based on molecular markers has apparent advantages since their genomic abundance, environment insensitivity, and non-tissue specific features. However, the limited number of available DNA markers, especially SSR markers, are insufficient to conduct related genetic diversity assessment studies in garlic.In this study,4372 EST-SSR markers were newly developed,and 12 polymorphic markers together with other 17 garlic SSR markers were used to assess the genetic diversity and population structure of 127 garlic accessions.The averaged polymorphism information content(PIC)of these 29 SSR markers was 0.36, ranging from 0.22 to 0.49. Seventy-nine polymorphic loci were detected among these accessions, with an average of 3.48 polymorphic loci per SSR. Both the clustering analyses based on either the genotype data of SSR markers or the phenotypic data of morphological traits obtained genetic distance divided the 127 garlic accessions into three clusters.Moreover,the Mantel test showed that genetic distance had no significant correlations with geographic distance, and weak correlations were found between genetic distance and the phenotypic traits. AMOVA analysis showed that the main genetic variation of this garlic germplasm collection existed in the within-population or cluster.Results of this study will be of great value for the genetic/breeding studies in garlic and enhance the utilization of these garlic germplasms.
Keywords:Garlic SSR markers Genetic diversity Population structure
1. Introduction
Garlic (Allium sativumL.), originated in central Asia, is one of the most worldwide cultivated and consumed horticultural crops since the ancient Egyptian period due to its edible and medicinal value (Vavilov, 1951; Hong and Etoh, 1996; Rahman and Lowe,2006). Although garlic is asexually propagated, it shows surprisingly high biodiversity, environmental adaptation capacity, and phenotypic plasticity (Volk et al., 2004). A large-scale diversity of different cultivars has been established in various cultivation areas(Bradley et al.,1996;Avato et al.,1998;Baghalian et al.,2005;Wang et al., 2014). These abundant garlic cultivars or germplasms can broaden the genetic variability and provide considerable opportunities for garlic genetics and breeding research (Zhao et al.,2011). The raw collections of different garlic cultivars, accumulated over the years, are usually classified based on phenotypic traits, which can easily lead to homonymy (same name for genetically different cultivars) and duplications or synonymy(same cultivars with different names) of garlic. That is especially problematic for the garlic with a similar appearance and significant phenotypic plasticity. Thus, how efficiently identify and distinguish of each garlic germplasms is of paramount importance to manage and maintain such genetic resources (Egea et al., 2017;Govindaraj et al., 2015).
The early identifications of garlic germplasms were mainly based on morphological characteristics that highly dependent on the field conditions and the local environments of their planting areas (Bradley et al., 1996; Al-Zahim et al., 1997). The genomic abundance, environment insensitivity, and non-tissue specific DNA markers showed significant advantages in biodiversity analysis of garlic and were gradually used in the identification and genetic diversity assessment of garlic accessions (Ovesn´a et al.,2014; Ipek et al., 2015). Furthermore, some DNA markers may be useful for marker-assisted selection in garlic breeding programs.Barboza et al. (2020) found markers AsESSR-30 and AsESSR-83 associated with flowering behavior, ecophysiological groups, and color types. The following types of markers including random amplified polymorphic DNA (RAPD) (Maaβ and Klaas, 1995),amplified fragment length polymorphism (AFLP) (Ipek et al.,2005), simple sequence repeats (SSR) (Cunha et al., 2014), and insertions-deletions (InDel) (Wang et al., 2016) were all successfully used in garlic germplasms evaluations. Among these DNA markers, SSR markers became the preferred ones since they are typically codominant, reproducible, cross-species transferable,and highly polymorphic.SSR markers can be classified as genomic(gSSR) and genic (EST-SSR) SSRs, which were developed with the sequences of genomic DNA and cDNA or expressed sequence tags(ESTs), respectively (Vaek et al., 2020). Compared with the gSSR,the development of EST-SSR markers is relatively inexpensive because the required sequence data of cDNA or EST are easily obtained from public databases(Liu et al.,2015).Besides,primers of EST-SSRs are designed from more conserved coding regions of the genome, which made the EST-SSR markers more useful and with higher cross-species transferability (Varshney et al., 2005).However, the EST-SSRs are usually less polymorphic than gSSRs,and their PCR amplified fragments may fluctuate as the presence of introns in flanking regions(Kalia et al.,2011),which may lead to that not all EST-SSRs are perfect SSR markers. Therefore, the development of sufficient EST-SSRs is the precondition for fully using this kind of DNA markers. Recently, large numbers of ESTSSR markers have been successfully developed for many plant species (Lu et al., 2013; Blair and Hurtado, 2013; Mohanty et al.,2013). However, due to the relatively large genome size (~16 Gb)and the few available fertile germplasms, the molecular and genetic studies of garlic were largely lagged than other vegetable crops (Meryem et al., 2015). In addition, the available number of SSR markers is also relatively limited in garlic(Cunha et al.,2012).Previously, fewer than 100 SSR markers were reported in garlic(Ma et al.,2009;Lee et al.,2011;Cunha et al.,2012).Until recently,Liu et al.(2015)developed 1506 SSR markers using EST(expressed sequence tags)derived from a transcriptome dataset.Considering the relatively large genome size of garlic, the available SSR markers are still insufficient for adequate genetic studies in garlic(Liu et al., 2015).
Overall,using the SSR markers to genotype garlic cultivars from the molecular DNA levels will be an efficient way for the identification of each garlic germplasms since the environment insensitivity and non-tissue specific advantages of DNA markers.However,the shortage of sufficiently high-quality SSR markers is a serious limitation for the DNA marker-based identification of garlics.Therefore, the first objective of this study is to develop more new SSR markers in garlic based on the EST sequences derived from our previous transcriptome data; and the other purpose is that using the newly designed SSR markers to distinguish the duplications or synonymy garlics and to evaluate the genetic diversity of a garlic germplasm collection including 127 accessions.
2. Materials and methods
2.1. Plant materials and morphological data collection
A total of 127 garlic accessions kept and propagated by the Vegetable Physiology and Biotechnology Laboratory, Northwest A&F University, were used for genetic diversity and population structure analysis. The detailed names and geographical origins of these garlics were listed in Table S1. These accessions were fieldgrown at the Wuquan Experiment Station (WES-NWAFU) of Northwest A&F University (108°08′E, 34°29′N) in Yangling,Shaanxi Province,China.These garlic accessions were grown in two growing seasons of 2019 and 2020, which were both sown in the September and harvested in April or May of the following year.About 500 individuals were planted for each garlic accession and the standard horticultural practices were performed following Liu et al. (2019). Thirty typical individuals were randomly selected for morphological data collection. The growth period of each garlic accession was recorded as the days from sowing to the harvest of mature bulbs. Garlic bulb related morphological traits, including bulb height(the height from the base of a bulb to the highest point),bulb width(maximum transverse width of a bulb),bulb weight(the average weight of 30 typical bulbs for each accession), rind color,and the clove number per bulb,were also recorded.All the collected phenotypic data were also listed in Table A.
2.2. Identification of SSR loci and marker development
We previously assembled and annotated 289,142 unigenes using a garlic transcriptome data set (Liu et al., 2020). Here, the simple sequence repeats or microsatellites (SSRs)were identified among these garlic unigenes using a computer program MISA(MIcroSAtellite identification tool) version 1.0. The default parameters were used to screen a minimum of 6 repeats for dinucleotide motifs and 5 repeats for trinucleotide, tetranucleotide,pentanucleotide, and hexanucleotide motifs. For those detected putative SSRs,the Primer 3.0 software was used to design flanking primers with the corresponding unigene sequences following the criteria used in Liu et al. (2015). To assess the qualities of these newly developed SSR primer pairs, fifty SSR primer pairs were randomly selected to evaluate the genetic diversity and population structure of a panel of garlic germplasms that includes 127 accessions collected worldwide. Besides, ten polymorphic garlic SSR markers previously used in our laboratory and seven SSR markers used by Ma et al. (2009) were also selected to genotype these garlic germplasms.The detailed sequence information of all SSR markers used and developed in this study are listed in Table 1 and Table S2.
2.3. DNA extraction, PCR amplification and genotyping
About 0.25 g freeze-dried young leaf samples were weighted and ground into a fine powder for each garlic accession. The total genomic DNA of each sample was extracted using the CTAB procedure (Murray and Thompson,1980). The quality and quantity of extracted DNAs were examined by electrophoresis in 1% agarose gel and measured using NanoDrop 2000 spectrophotometers,respectively.The high-quality DNAs were diluted into 50 ng/μl with ddH2O and stored at -20°C until further use.
PCR amplifications were carried in 10 μl reactions, each containing 2 μl template DNA (50 ng/μl), 0.5 μl forward primers(5 μmol/l), 0.5 μl reverse primers (5 μmol/l), 2 μl ddH2O and 5 μl 2 × Taq PCR Master mix(Tiangen Biotech Co.,LTD,Beijing, China).The PCR program was as follows:5 min at 95°C,40 s denaturing at 94°C,40 s annealing at 68°C and 40 s elongation at 72°C,followed by a 2°C reduction in the annealing temperature per cycle for 6 cycles.Then reduce annealing temperature in each cycle by 1°C for 8 cycles from 58°C;the annealing temperature was maintained at 50°C for the remaining 20 cycles,followed by a final step at 72°C for 5 min. The amplified PCR products were separated by vertical electrophoresis on 8% polyacrylamide gel in 1 × TBE buffer at a constant 180 V for 1 h, visualized with silver staining, and photographed with a digital camera.The clear and unambiguous bands of all the polymorphic SSR markers were scored for 127 garlic accessions and calculated into co-dominant genotypic matrix in GeneAlEx6.5 (Peakall and Smouse, 2012) following Zhu et al.(2016), which was used for the following data analysis.
2.4. Data analysis
The ANOVA of phenotypic traits between the years 2019 and 2020 was conducted first with SPSS 21, and then the ‘pheatmap’Rpackage was used to conduct a phenotypic cluster analysis of the 127 garlic accessions. In addition, the ‘pairs. Panels’ function in‘psych’Rpackage(Northwestern University,Evanston,IL,USA)was used for analyzing the correlations among phenotypic traits(https://cran.r-project.org/web/packages/psych/citation.html).
General statistical parameters that reflect the genetic diversity of the 127 garlic accessions, according to the summarization of Pagnotta (2018), the number of alleles (Na), expected heterozygosity(He),and Nei's gene diversity(H)were determined using the software of Popgene (v.1.32). The polymorphism information content(PIC) was calculated with PowerMarker (v.3.25). The pairwise genetic distances were used to examine the hierarchical clustering of garlic accessions via a dendrogram based on the unweighted pair-group method with an arithmetic mean (UPGMA) analyses and the neighbor joining tree was constructed by NTSYS 2.10 software.
The GenAlEx 6.5 was used to estimate the analysis of molecular variance(AMOVA).The redundant garlic accessions were identified by genetic distance(GD=0),and the GD was calculated with NTSYS 2.10 software. Further analysis was performed using 102 garlic accessions after removing the redundancy.
For population structure analysis, the Bayesian model-based clustering was performed in STRUCTURE v.2.3.4 (Pritchard et al.,2000) to infer the appropriate clusters (K), using a burn-in of 10,000,run length of 150,000,and assuming admixture model and correlated allele frequencies. Ten runs of STRUCTURE were performed by setting the number of populations(K)from 1 to 10.The most probable value of K was select by ΔK methodology using the web-based software STRUCTURE HARVESTER v.0.6.94 (Earl and VonHoldt, 2012).
The correlation between the cophenetic matrix of Euclidean distances (morphological traits) and the cophenetic matrix of genetic distances was also calculated with GenAlEx 6.5 through the Mantel test. The comparison of morphological traits among different cluster accessions was conducted with ANOVA used SPSS 21, in which theP-value= 0.05 was used as a cut-off for the significant difference.
3. Results
3.1. The diversity of 127 garlic accessions revealed by the investigated morphological traits
Morphological traits, including growth period, cloves number,bulb height,width,weight, and rind color of 127 garlic accessions,were listed in Table S1. The ANOVA showed no significant differences for these observed phenotypic traits between the two growing seasons of 2019 and 2020 (Table S3).
Based on the observed data of 2020, this garlic germplasm collection had a relatively higher diversity among the investigated traits(Table S1.and Fig.1).For the rind color of garlic bulbs,eightytwo accessions had purple rinds, while all the rest showed white rinds. Among the 127 garlic accessions, bulb height ranged from 24.4 mm(GS018 and GS112)to 45.0 mm(GS025),with an average height of 33.3 mm. Both GS018 and GS025 showed the narrowest(28.4 mm)and widest(62.7 mm)bulb width,respectively,with the mean bulb width of 43.8 mm.The minimum clove number was 4.7 for GS088,while the highest was 16.0 for GS024,with an average of 11. The bulb weight exhibited the largest variation ranging from 7.2 g for GS023 and GS111 to 70.1 g for GS024.The earliest-mature accession was GS044 with 246 days growth period, while the latest-harvest garlics were at 290 days for GS011, GS012, GS013,GS014, GS016, GS035, GS083, and GS118. In addition, for each accession, the bulb width was larger than the bulb length, which suggested that all these garlic accessions are characterized with flat shape bulbs.
Except for the rind color, the distributions and Pearson correlations of these investigated traits were calculated and displayed in Fig. 2. As shown in Fig. 2, the growth period, clove number, bulb height, width, and weight were all continuously distributed over the 127 accessions. The distributions of most accessions were within the ranges of 32-38 mm for bulb height,35-50 mm for bulb width, 10-14 for clove number, 20-40 g for bulb weight, and 260-270 days for growth period. According to the calculated correlation coefficients, bulb weight had significant positive correlations with bulb height,width,and clove number,especially for the bulb width, which had a very strong correlation. Besides, clove number, bulb height, and width showed significant positive correlations with each other.However,growth period only had a weak correlation with bulb height.

Fig.2. Pearson correlation coefficients among phenotypic traits in 127 garlic accessions. NS,
Clustering analysis of these 127 garlic accessions was performed based on the investigated morphological traits. The phenotypicbased dendrogram (Fig. 3) illustrates that all the garlic accessions were classified into three major groups (Cluster I, II, and III). The majority(116 accessions)of this garlic collection were grouped into Cluster III,with only three accessions included in Cluster I,of which two (GS024 and GS025) were from Belgium, and one (GS120) is from the Xinjiang Province of China, and the other 8 accessions were grouped as Cluster II, which from America (GS018), Japan(GS022),Thailand(GS023)and China(GS077,GS088,GS108,GS111,GS112).
3.2. Distribution of SSR motifs and primer pairs development
A total of 4372 SSR loci were detected from assembled sequences of 289,142 unigenes. The frequencies, types, and distributions of these SSRs were analyzed and shown in Fig.4.For these 4372 SSR loci, the dinucleotide repeat motifs were the most abundant type (2,461, representing 56.3%), followed by trinucleotide repeat motifs (1,811, representing 41.4%) and tetranucleotide repeat motifs (91, representing 2.1%), while the distributions of pent nucleotide and hexanucleotide repeat motifs were relatively rare(9,representing 0.2%).In addition,the repeat times of different SSR motifs were mainly scattered from five to ten. Among these identified SSR loci,83 motifs were detected,in which nearly half of these SSRs were derived from five types of dinucleotide repeat motifs, including TA/TA (893, representing 20.4%), TG/CA (551,representing 12.6%), AC/GT (516, representing 11.8%), AG/CT (246,representing 5.6%), and TC/GA (227, representing 5.2%).
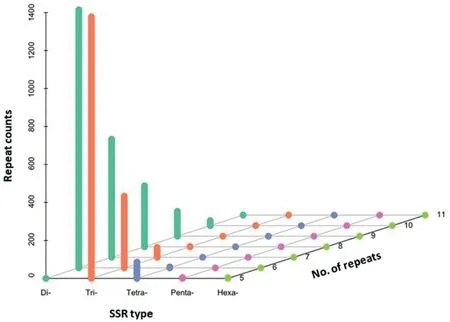
Fig.4. Distribution of various SSR motifs with different numbers in transcriptome of garlic.
The designed primer pairs of these 4372 SSRs are listed in the Table S2.The putative amplified product sizes of these SSR markers ranged from 100 to 280 bp with a mean size of 210.2 bp. These primers had the following features: the sequence lengths ranging from 18 to 27 bp,the GC contents varied from 25.9 to 77.8%,and the Tm changed from 57.0 to 62.9°C.The averaged sequence length,GC content, and Tm of these SSR primers were 20.6 bp, 50.8%, and 59.6°C, respectively. Furthermore, compared with the garlic SSR markers developed by Liu et al. (2015), 244 consistent markers were found among the markers of this study (Table S2). This may suggest that most SSR markers of the present work are newly developed markers for garlic.
3.3. SSR marker assay and their informativeness
To assess the qualities of the newly developed SSR markers,50 pairs of SSR primers were randomly selected to firstly genotype 10 of the 127 garlic accessions.Among these 50 SSR primer pairs,39 SSR markers (78.0%) successfully amplified targeted DNA fragments, of which 12 SSR primer pairs exhibited polymorphism among these 10 selected garlic accessions. These 12 markers and another 17 SSR markers listed in Table 1 were used together to genotype all the 127 garlic accessions. The genotyping analysis indicated a total of 79 alleles among these 127 accessions, with a range of 2-6 alleles and an average of 3.48 alleles per SSR marker(Table 1). Furthermore, the polymorphism information of SSR markers reflected by parameters expected heterozygosity (He),Nei's gene diversity (H), and the polymorphism information content (PIC) were also calculated and listed in Table 1. For these 29 markers,theHeranged from 0.01 to 0.95 with a mean value of 0.67. TheHof each marker changed from 0.01 to 0.47, with the averagedH= 0.25. The PIC varied from 0.22 to 0.49, with an average of 0.36, which indicated that these SSR markers were good enough for evaluating the genetic diversity of the 127 garlic accessions.

Fig.1. Typical garlic bulbs (a) and cloves (b) with diverse phenotypes.
3.4. The genetic diversity and population structure of the 127 garlic accessions
The genetic distance matrix of 127 garlic accessions was calculated based on the genotyping data of all 79 alleles. The neighborjoining tree of these accessions was constructed with the obtained genetic distances by the UPGMA method,and the dendrogram was shown in Fig. 5. These 127 accessions were mainly grouped into three clusters,in which Cluster 1,2,and 3 had 7,15,and 105 garlic accessions,respectively.No direct relations were revealed between the geographical location of each of the garlic genotypes and their origin area. Although the garlic genotypes in Cluster 1 were all collected in China,they originated from entirely different locations of China, e.g. four accessions (GS081, GS083, GS113, and GS118)were collected from the northwest of China,one(GS076)from the southwest of China,and the other two(GS087 and GS090)from the northeast and north of China, respectively. Similarly, the original locations of accessions in Cluster 2 were scattered at the east,southwest, central, north, and northwest parts of China, respectively. In Cluster 3, 78 garlic accessions were collected from different regions of China, and the other 27 genotypes were obtained from other 15 countries.
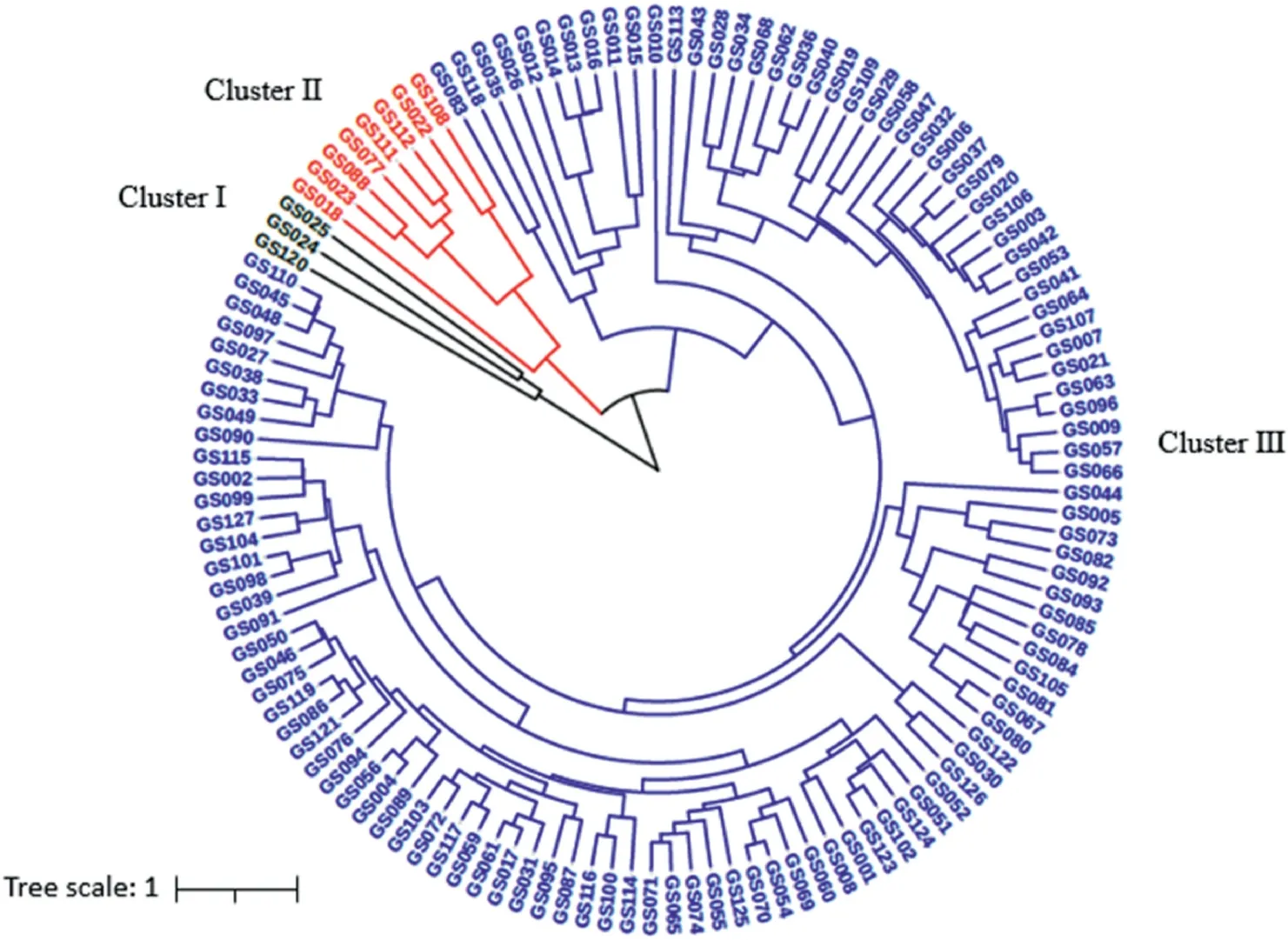
Fig. 3. Cluster dendrogram of 127 garlic germplasm based on phenotypic traits.
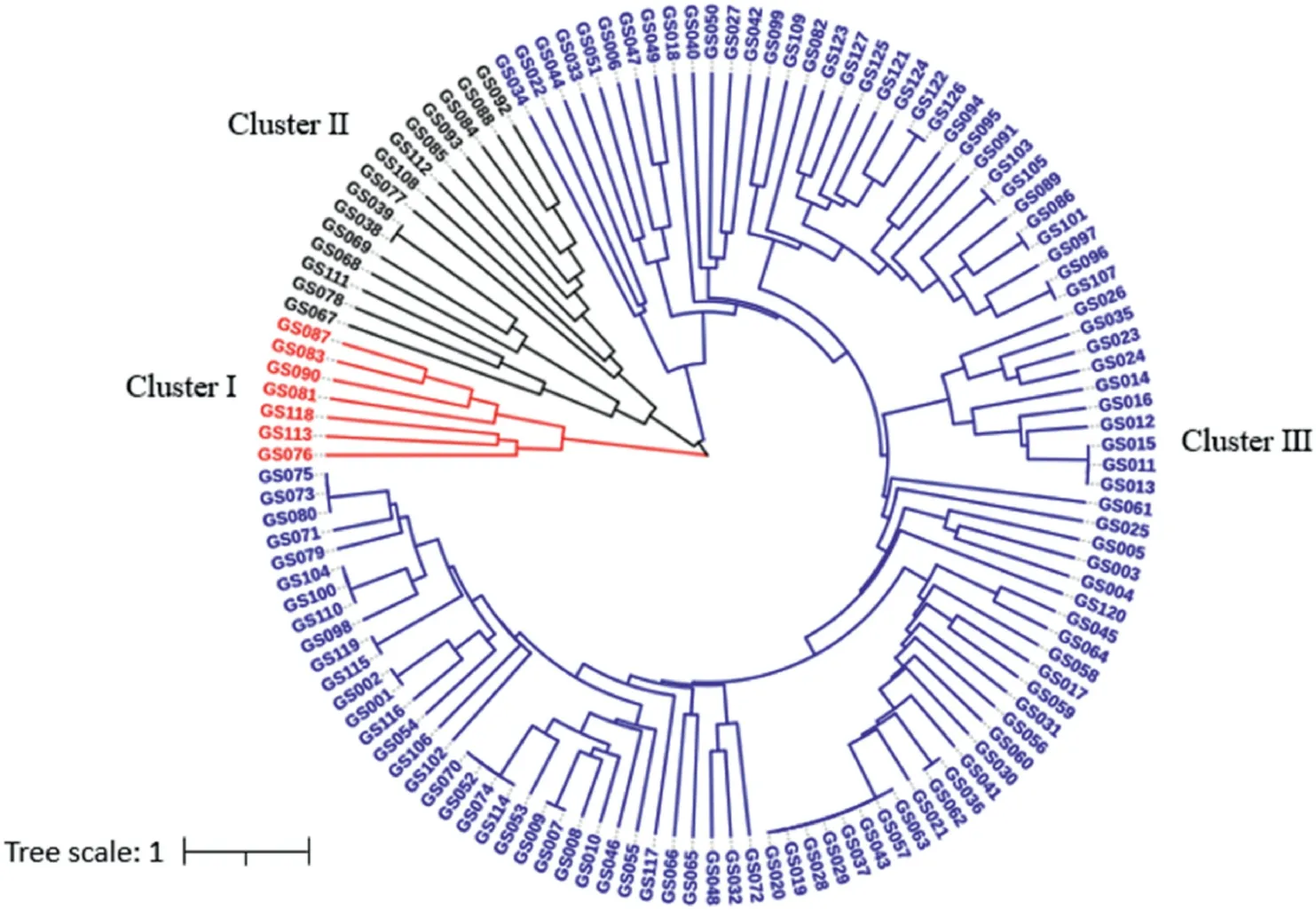
Fig. 5. UPGMA dendrogram demonstrating genetic relationships among 127 garlic accessions.
For further study of population structure and genetic variation,a total of 14 groups of potential redundancy garlic genotypes were identified from the 127 garlic accessions according to the genetic distance(GD)=0 and the SSR marker based dendrogram(Table 2).Group 4 had the highest number (8 garlic accessions) of redundancy genotypes, followed by Group 7, which had 4 garlic genotypes; three repetitive garlic genotypes were found for all of the Group 3, 8, and 11; while two redundancy garlic accessions per group were identified for all the rest 9 Groups. Although some redundancy of garlic accessions had the same geographical origins,the redundancy genotypes originated from different provinces of China or from different countries were also common, such as the repetitive genotypes of GS001 vs GS002,GS007 vs GS009,GS011 vs GS013 vs GS015, and so forth. Overall, after eliminating these redundancy genotypes (25 accessions), 102 unique garlic germplasms were determined from the raw 127 garlic accessions.

Table 2 The redundant garlic accessions identified by the genetic distance.
After removing the 25 redundant accessions, the genetic population structure of the unique 102 garlic genotypes was further analyzed based on their genotypic data derived from 29 SSR markers. Analyses of K-value (number of clusters) from 1 to 10 revealed the highest peak of ΔK at K = 3, where there was no tendency to divide into subgroups (Fig. S1), which indicated that three clusters were involved in this garlic collection of 102 accessions.The result of genetic structure clearly showed three clusters,including Cluster 1,2,and 3.For each cluster,most of the belonged accessions were from a single primitive ancestor with a few mixed individuals(Fig.6).Furthermore,the structure distribution of three clusters of these 102 garlic accessions was highly consistent with the cluster analysis of the whole collection of 127 garlic, in which the Cluster 1, 2, and 3 of the genetic structure analyses corresponded to the Cluster I, II, and III of the UPGMA-based dendrogram, respectively.
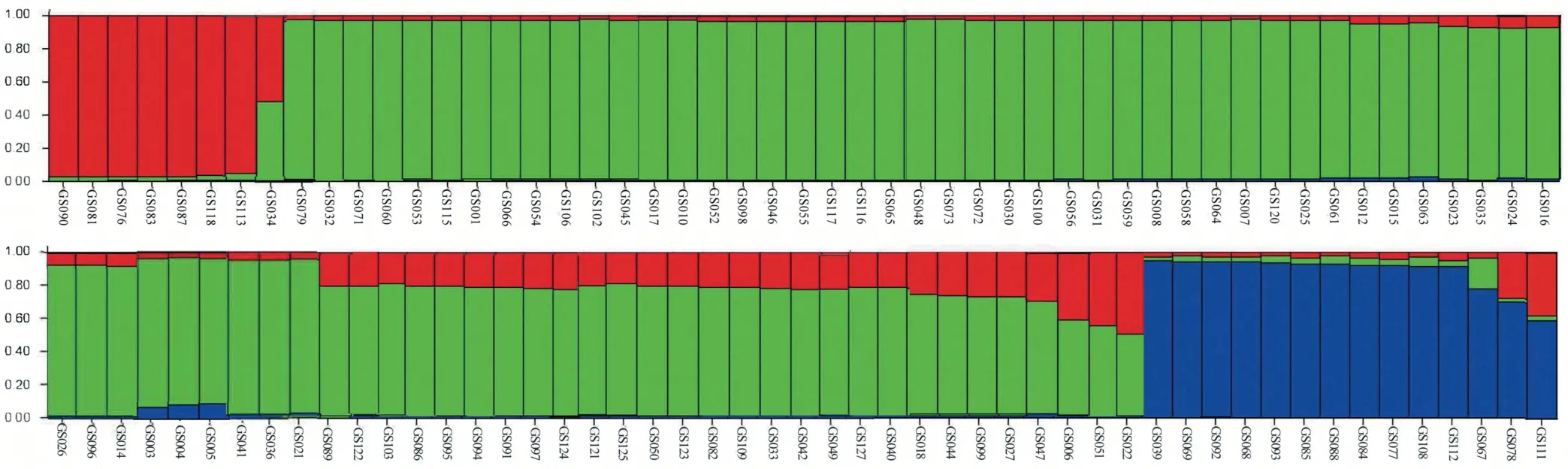
Fig. 6. Genetic structure of 102 garlic accessions as inferred by STRUCTURE based on 29 SSRs.
The population differentiation was evaluated through the analysis of molecular variance(AMOVA)utilizing the obtained data of SSR markers,in which the genetic variation of within-population was 90%,and that of among populations was only 10%(Table 3).In addition,the overallFstvalue(0.18)indicated a high level of genetic differentiation among the collection of 102 garlic accessions, according to Wright(1978)who defined the genetic differentiation as low forFst< 0.05, moderate for 0.05 <Fst<0.15, high for 0.15 <Fst<0.25, and very high forFst>0.25.
3.5. The relationship between the genetic differentiation and morphological traits of garlics
The correlations between genetic distance and phenotypic traits were analyzed by the Mantel test (Table 4). The genetic distance had weak correlations with all the investigated morphological traits, in which the growth period (r= 0.13) showed the lowest correlation with genetic distance, while the correlation between the clove number and the genetic distance was the highest(r= 0.37). The correlation coefficients of bulb weight, height, and width were 0.16, 0.21, and 0.15, respectively. In addition, the potential correlation between the genetic distance and geographical distance was also analyzed, while no significant correlation was found for these 102 garlic accessions.
The potential differences of morphological traits among the three clusters were further analyzed (Table 5). The results showed that all the traits of Cluster 3 were significantly higher than those of Cluster 2,except for clove number.The traits of Cluster 1 were also significantly higher than those of Cluster 2,when compared Cluster 1 and Cluster 3. Only the clove number showed a significant difference,where the accessions of Cluster 3 had more cloves per bulb.Overall,both the Cluster 1 and Cluster 3 were relatively superior to the Cluster 2 in phenotypic traits. Besides, when considering the rind colors of garlic bulbs,all these three clusters did not show any specificity since each cluster scattered with both white and purple rind colors.

Table 3 Analysis of molecular variance (AMOVA) of 102 garlic accessions.

Table 4 The correlations between the genetic distance and geographical distance and phenotypic traits based on the Mantel test.

Table 5 ANOVA of phenotypic traits and growth period in different clusters.
4. Discussion
Although most garlic varieties propagate asexually,they exhibit considerable morphological differences within and between them(Bradley et al.,1996). Evaluating the genetic variation of cultivated garlic accessions is helpful for phenotypic identification and core germplasm construction. In addition, the analysis of genetic diversity and relatedness between accessions are also important for garlic selection and breeding purposes (Figliuolo et al., 2001). In early studies, the variation of different garlic clones was mainly investigated in morphology and enzymology(Al-Zahim et al.,1997;Ipek et al., 2003). Highly morphological diversity was observed in garlic from several phenotypic traits,including the bulb size,shape and rind color,clove rind color,bolting habit,and clove number and weight(Singh et al.,2014;Raja et al.,2017).In the current study,the 127 garlic accessions exhibited relatively higher diversity among the investigated traits where the clustering analysis divided these accessions into three clusters. However, based on the morphological traits only, the potential redundant accessions were not accurately identified, which might indicate the shortage of genetic diversity evaluation of garlic germplasms from the aspect of morphology.Polyzos et al.(2019)reported that the garlic diversity examined based on morphological traits is much dependent on the genetic composition and the environmental conditions such as cultivation practices, soil properties, and fertilizing regimes. This might explain the major reasons for the disadvantage of morphological traits based genetic diversity analysis in garlic.
For genetic diversity evaluation,SSR markers have been widely applied in many plant species to evaluate genetic diversity, to construct genetic maps, and to determine species lineages. However,the insufficient number of SSR markers is a major obstacle for the related genetic studies of garlic.In the current study,4372 SSR markers were newly developed from the sequences of 289,142 garlic unigenes that were assembled from our previous transcriptome data (Liu et al., 2020). Among these SSR markers, the dinucleotide repeat motifs were the most abundant type,followed by trinucleotide repeat motifs and tetranucleotide repeat motif.This is different from the results of Liu et al. (2015), who reported that the trinucleotide repeat motifs were the most abundant type,followed by the dinucleotide repeat motifs. This might be mainly due to the SSR search criteria, the size of the dataset, and the database-mining tools (Varshney et al., 2005; Aggarwal et al.,2007). Besides, there were only 244 consistent SSR markers shared between Liu et al. (2015) and those reported here, which might also verify the potential differences of the assembled unigene sequences of these two studies.To check the qualities of these newly developed SSR markers, 50 pairs of SSR primers were randomly selected to genotype 10 garlic accessions, in which 78%markers (39) successfully amplified target bands, and the remaining 22%SSR markers(12)did not amplify any fragments.The failed amplifications of these SSRs may be probably due to the fact that the primers were designed across splice sites or large introns(Varshney et al., 2006; Cloutier et al.,2009; Liu et al., 2013).
Twelve polymorphic markers from those 39 newly developed and verified SSR markers were used to evaluate the genetic diversity of 127 garlic accessions together with another 10 EST ESTSSR markers previously developed by our lab and 7 SSR markers reported by Ma et al.(2009).The averaged PIC value of these 29 SSR markers was 0.36 with a range from 0.22 to 0.49,which is similar to the mean PIC value(0.38,ranging from 0.30 to 0.54)of 10 EST-SSRs used by Barboza et al.(2020)for assessing the genetic diversity of a collection of 73 garlic accessions. However, Ipek et al. (2015) obtained the mean PIC value of 0.60 for 26 EST-SSRs used in 31 garlic accessions. A higher mean PIC value in Ipek et al. (2015) probably caused by the 31 garlic were intentionally selected based on their previous AFLP analysis work (Ipek et al., 2003) to maximize the genetic variations,whereas no pre-selection and classification were performed for the 127 accessions used in the present research. In addition,the numbers or types of used markers and the population size or the actual genetic variation of an evaluated germplasm collection might all contribute to the changes of the PIC values(Barboza et al., 2020).
When assessing the genetic diversity and population structure of a germplasm collection,the SSR or other DNA markers were able to overcome the problem in the nomenclature of garlic accessions and the redundancy. The guaranteed identification of potential duplicates is helpful for reducing costs in the maintenance of garlic germplasms. Based on the genetic distance and the SSR markerbased dendrogram, 25 potential duplicates were identified among the 127 garlic accessions (Table 3). These redundancy garlic accessions may be due to the farmers in different areas tend to exchange their garlic varieties/produce from year to year, to protect yield against degeneration caused by continuous cropping using the same variety, and each farmer might name the same garlic variety with obviously different names;thus,some different garlic varieties were improperly given a same name(Wang et al.,2016).In addition, both the cluster analysis of 127 accessions and the population structure analysis of 102 unique garlic accessions revealed that three major clusters existed for the garlic germplasms used in this study. However, the garlic accessions that grouped into the same cluster were not completely consistent with their geographical origins. Moreover, the Mantel test results also verified that there was no significant correlation between genetic distance and geographic distance, which was also found by Ipek et al. (2003),Pooler and Simon (1993), Wang et al. (2016), and Morales et al.(2013). Furthermore, many of the garlic accessions may have secondary source data rather than original wild collection data associated with them, which makes it difficult to trace their geographical origins (Volk et al., 2004). All the above issues might lead to the high redundancy accessions among the worldwide collections of garlic germplasms. In the present study, AMOVA analysis indicated 90% of the variation due to differences withinpopulation variation, and only 10% of the variation was due to differences among populations,which might suggest the presence of genetic structures. This was consistent with the results of Zhao et al. (2011), who observed that 84.4% variation was from withinpopulation differences and 15.6% variation was due to the divergences between populations. However, the results of Barboza et al. (2020) showed that the variation within-population and among populations was 72% and 28%, respectively. The proportional difference of variation sources might have been caused by the significantly different garlic accessions used in each of the related studies. In addition,for the 102 unique garlic accessions of this study,a higher level of genetic differentiation was indicated by the overallFstvalue (0.18), which might suggest that these accessions are valuable for future breeding of new garlic cultivars.
Through the ages, garlic is asexually propagated by using their cloves since the lack of fertile and seed setting germplasms,which leads to that the introduction is the only effective way for obtaining relatively good varieties in the cultivation of garlic. The extensive and frequent introduction of garlic cultivars from different areas results with serious homonym and synonym issues for garlic accessions.Efficiently analysis of the genetic diversity and relatedness of different garlic cultivars, using the SSR or other molecular markers from the genomic DNA level, is of important for identification of potential redundant accessions and construction of core garlic germplasm panels, which could further reducing the maintenance cost of garlic germplasms and promoting the selection and breeding of high-quality garlic cultivars. Here, in this study, 25 duplicated cultivars were successfully identified from 127 garlic accessions based on the genotyping matrix of 29 SSR markers,which indicates the effectiveness and advantages of SSR markers in the identification of garlic germplasms. The relatively higher redundant garlic cultivars (19.68%, 25 of the 127 accessions) identified in this work also further confirmed the universal phenomena of homonym and synonym in garlic.Until now,the relatively large genome size(~16 Gb)and few available fertile germplasms are still restricting the processes of the molecular and genetic studies in garlic (Meryem et al., 2015). Although we newly developed more than 4000 SSR markers, given the large genome size of garlic, the available molecular DNA markers are still not enough. The newly released garlic genome might provide good opportunity for identifying and developing of new DNA markers from the genome-wide level.Then,thousands of new SSR or SNP markers will be available for using in the genetic studies of garlic. Anyway, at the present stage, the newly developed SSR markers and the evaluated nonredundant garlic accessions are still valuable for promoting the related genetic studies in garlic and for the researchers, breeders,and producers to further utilize these garlic germplasms.
5. Conclusion
In conclusion, 4372 EST-SSRs were newly developed from assembled unigene sequences for garlic, in which 12 polymorphic markers combined with other 17 SSR markers were successfully used for the genetic diversity and population structure analysis of a garlic germplasm collection.There were 25 duplications among the evaluated 127 garlic accessions, and the rest of 102 unique garlic accessions can be divided into three clusters. Among this garlic germplasm collection, some interdependent traits such as bulb weight clove number, bulb height, and width exhibited significant positive correlations with each other.Overall,the newly developed SSR markers will be of great value for the related genetic studies in garlic, and the assessment of genetic diversity and population structure of the involved garlic accessions will also provide valuable information for further utilizing these garlic germplasms.
Author contributions
X.Li conducted the majority of the reported research.L.Qiao and B. Chen helped with genotyping of SSR markers. Y. Zheng,C. Zhi, and S. Zhang helped with the morphological data collection. Y. Pan and Z. Cheng conceived and supervised the research. X. Li and Y. Pan wrote the manuscript with the input of Z. Cheng. All authors reviewed and approved the final submission.
Declaration of competing interest
None.
Acknowledgments
This work was supported by the Education Development Fund of Northwest A&F University (2017) to Z. Cheng and the Chinese Universities Scientific Fund(2452019017) to Y. Pan.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.pld.2021.08.001.

- 植物多样性的其它文章
- GUIDE FOR AUTHORS
- Taxonomic synopsis of Berberis (Berberidaceae) from the northern Hengduan mountains region in China,with descriptions of seven new species
- Plant invasions facilitated by suppression of root nutrient acquisition rather than by disruption of mycorrhizal association in the nativeplant
- Evolutionary importance of the relationship between cytogeography and climate: New insights on creosote bushes from North and South America
- Mapping the habitat suitability of Ottelia species in Africa
- Adaptive responses drive the success of polyploid yellowcresses(Rorippa, Brassicaceae) in the Hengduan Mountains, a temperate biodiversity hotspot