
超高效液相色谱-串联质谱测定米粉中米酵菌酸
2022-10-10 01:56于浩洋冯静李颜岩
化学分析计量 2022年9期
于浩洋,冯静,李颜岩
(辽宁省疾病预防控制中心,沈阳 110005)
2020 年,黑龙江省鸡西市鸡东县某家庭聚餐导致多人中毒事件引发关注[1]。经调查发现,在该家庭聚餐食品玉米面中检出了高浓度的米酵菌酸,同时在患者胃液中亦有检出。1953 年以来,我国学者陆续在16 个省份发现了由当地特色食品引起的椰假单胞菌食物中毒545 起,中毒人数3 352 人,死亡1 401 人,平均病死率高达41.80%,是我国病死率较高的一种微生物性食物中毒[2]。
米酵菌酸是椰毒假单胞菌酵米面亚种产生的一种可以引起食物中毒的毒素,具有强烈的毒性[3]。谷类制品(米粉、发酵玉米面等)、变质银耳和薯类制品(马铃薯粉条、甘薯淀粉、山芋淀粉等)为主要受污染食品[4]。米酵菌酸无臭无味,被污染的食品没有明显变化[5],一旦上述食品因储存不当而产生米酵菌酸毒素,即使加热烹制也无法消除。该菌产生的毒素米酵菌酸对人体的肝、肾、心、脑等重要器官均能产生严重的损害,是引起人们严重食物中毒和死亡的主要原因[6]。据文献报道[7],米酵菌酸中毒潜伏期仅为0.5~2 h,病情发展迅速,很快出现肝、肾等多脏器实质性损伤[5]。建立米酵菌酸的快速检测方法,对及时鉴定中毒病因、中毒患者的毒物溯源、评估中毒水平以及中毒患者的救治工作具有重要意义。
目前,针对米酵菌酸常用的检测方法包括液相色谱法[8-10]、液相色谱-质谱联用法[11-14]。现行的食品安全国家标准GB 5009.189—2016[15]采用高效液相色谱法测定发酵米面及其制品、银耳及其制品、等食品中的米酵菌酸。液相色谱法具有灵敏度较低且过程繁琐耗时、样品处理试剂消耗量大等缺点。覃冬杰等[16]采用超高效液相色谱-串联质谱法测定柳州螺蛳粉中米酵菌酸,虽大大提高了方法的灵敏度、减少了试剂的使用量,但其前处理采用MAX固相萃取柱净化,耗时较长。
笔者采用超高效液相色谱-串联质谱(HPLCMS/MS)法建立一种快速测定米粉中米酵菌酸的分析方法,该方法分析速度快,样品处理简单高效,定性、定量准确,检出限低,灵敏度高,重现性好,是快速、准确测定米粉中米酵菌酸的可靠方法。
1 实验部分
1.1 主要仪器与试剂
超高效液相色谱-串联质谱仪:3200QTRAP型,美国AB 公司。
涡旋振荡器:Lab Dancer 型,德国艾卡公司。
高速冷冻离心机:Thermo ST16R 型,美国赛默飞世尔公司。
电子天平:BS 110S 型,感量为0.1 mg,德国赛多利斯公司。
超纯水系统:MILLI-Q Gradient 型,法国默克密理博公司。
超声波清洗器:KQ5200DB 型,昆山市超声仪器有限公司。
针式滤器:Pall Corpration Acrodisc 型,孔径为0.2 μm,直径为13 mm,美国颇尔公司。
水中米酵菌酸溶液:10 μg/mL,生产批号为S057505,天津阿尔塔科技有限公司。乙腈、甲醇:色谱纯,美国费希尔公司。氨水:分析纯,西陇科学股份有限公司。实验用水为超纯水。
1.2 仪器工作条件
1.2.1 色谱
色谱柱:ACQUITY UPLC HSS T3 柱(100 mm×2.1 mm,1.8 μm,美国沃特世公司);进样体积:10 μL;柱温:40 ℃;样品室温度:4 ℃;流动相:A 相为水,B 相为乙腈,流量为0.4 mL/min,梯度洗脱;梯度洗脱程序:0~1.0 min 80% A,1.0~2.0 min 80%~5% A,2.0~4.0 min 5% A,4.0~4.1 min 5%~80% A,4.1~6.0 min 80% A。
1.2.2 串联质谱
离子源:电喷雾离子源(ESI);扫描方式:负离子扫描;离子喷雾电压:- 4 500 V;离子源温度:550 ℃;喷雾气压力:413.7 kPa;辅助加热器压力:413.7 kPa;气帘气压力:137.9 kPa;其它质谱参数及保留时间见表1。
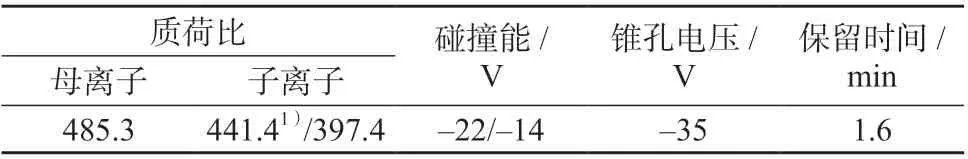
表1 米酵菌酸的质谱参数及保留时间
1.3 实验步骤
1.3.1 样品预处理
准确称取1 g 米粉(精确至0.01 g),置于50 mL离心管中,加入15 mL 甲醇-氨水溶液(每80 mL甲醇加入1 mL 氨水,加水定容至100 mL),涡旋提取2 min,于室温下置于暗处浸泡1 h,超声提取30 min,以10 000 r/min 离心5 min,过0.22 μm 滤膜后上机分析。
1.3.2 溶液配制
米酵菌酸标准溶液:60 μg/mL,准确吸取水中米酵菌酸溶液适量,用甲醇配制而得。
米酵菌酸标准储备液:3.0 μg/mL,准确吸取米酵菌酸标准溶液500 μL 于10 mL 容量瓶中,用甲醇定容至标线,于-20 ℃下保存。
系列米酵菌酸标准工作溶液:取适量米酵菌酸标准储备液于空白样品中,按照1.3.1 方式进行处理,配制成米酵菌酸质量浓度分别为2、10、50、100、150、200 ng/mL 的系列标准工作溶液。
2 结果与讨论
2.1 质谱条件优化
米酵菌酸是含有3 个羧基的脂肪酸,容易失去质子带上负电荷[17],故选择ESI 负离子模式检测。在负离子模式下,优化质谱条件,发现Q1 扫描时米酵菌酸失去1 个质子形成了丰度较高的[M-H]-峰,其质荷比为485.3;选择[M-H]-峰作为其母离子进行MS2 扫描,找到了丰度较大稳定性较好的两个子离子,分别为m/z441.4、m/z397.4,此二碎片离子分别为母离子失去一分子CO2和两分子CO2所形成。再采用MRM 模式优化碰撞能、锥孔电压等参数,使分子离子对的响应值达到最强,其中,m/z441.4 离子响应较强,作为定量离子;m/z397.4 离子作为定性离子。
2.2 色谱条件优化
在相同的实验条件下,分别比较了乙腈-水、乙腈-0.1%甲酸水、乙腈-0.1%氨水三种流动相对米酵菌酸色谱峰峰形及响应值的影响。由于米酵菌酸采用负离子扫描模式进行检测,理论上碱性流动相会促进米酵菌酸电离,增强米酵菌酸的响应。然而试验结果表明,在碱性环境下,米酵菌酸的响应值偏低、色谱峰形不佳且拖尾明显。
有文献报道提出,在酸性环境下,米酵菌酸具有较好的峰形和较高的响应值[18-19]。试验发现,以乙腈-0.1%甲酸水为流动相时,米酵菌酸响应值比碱性环境虽有提高,但色谱峰有明显分叉;以乙腈-水作为流动相时,米酵菌酸色谱峰半峰宽、峰形及响应值与酸性及碱性环境相比有明显改善,因此选用乙腈-水作为流动相。
以乙腈-水为流动相,采用ACQUITY UPLC HSS T3 色谱柱对米酵菌酸进行检测,结果表明,与等度洗脱相比,梯度洗脱分析时间更短、色谱峰半峰宽更窄,并且能克服等度洗脱时待测物与干扰杂质一起流出、对待测物的离子化产生抑制作用以及色谱峰背景干扰的缺点。优化后的梯度洗脱程序见1.2.1。
2.3 样品处理条件优化
2.3.1 萃取
米酵菌酸是一种脂溶性的酸性化合物,易溶于乙醚、甲醇、氯仿、乙腈等有机溶剂和碱性水溶液[20]。文献[16]表明以甲醇和乙腈作为提取溶剂时米酵菌酸回收率无明显差别,使用氯仿作萃取剂则回收率较低,从毒性及经济性考虑,采用甲醇作为提取溶剂。实验比较了甲醇、80%甲醇-水和50%甲醇-水对米酵菌酸提取效率的影响,结果表明,80%甲醇-水对米酵菌酸提取率最高。由于米酵菌酸分子结构中含有3 个羧基,在碱性环境下能够解离出氢离子而带一部分负电荷,更易溶于有机溶剂中,因此考虑在80%甲醇-水溶液中加入一定量的氨水,最终确定选择体积分数为80%的甲醇水溶液(含1%氨水)作为提取液。
2.3.2 超声时间
超声提取具有提取效率高、提取时间短、操作简单等优点[21],成为近年来应用较为广泛的食品提取方式之一,试验考察了超声提取时间分别为10、20、30、40 min 时米酵菌酸的提取效率,发现超声时间为30 min 时回收率最高,延长超声时间对增加回收率无明显影响,因此选择超声时间为30 min。
2.4 方法的线性关系、检出限与定量限
在最优实验条件下测试系列米酵菌酸标准工作溶液,以溶液的质量浓度(x)为横坐标,定量离子对色谱峰面积(y)为纵坐标,进行线性回归,计算线性方程、相关系数。
以3 倍信噪比对应的质量浓度作为方法的检出限,以10 倍信噪比对应的质量浓度作为方法的定量限。
本法测定米酵菌酸的线性范围、线性方程、相关系数、检出限和定量限列于表2。由表2 可知,米酵菌酸的质量浓度在2~200 μg/L 范围内线性关系良好,相关系数为0.998。检出限为0.67 μg/L,定量限为2.0 μg/L。

表2 米酵菌酸的线性范围、线性方程、相关系数、检出限与定量限
2.5 加标回收试验
准确称取18 份空白米粉1 g 作为本底,分别加入高、中、低三个质量浓度水平的米酵菌酸,每个浓度水平进行6 次平行测定,试验结果见表3。由表3 可知,米酵菌酸的平均加标回收率为96.1%~98.4%,测定结果的相对标准偏差为0.7%~2.5%,说明该方法具有较好的精密度和较高的准确度。
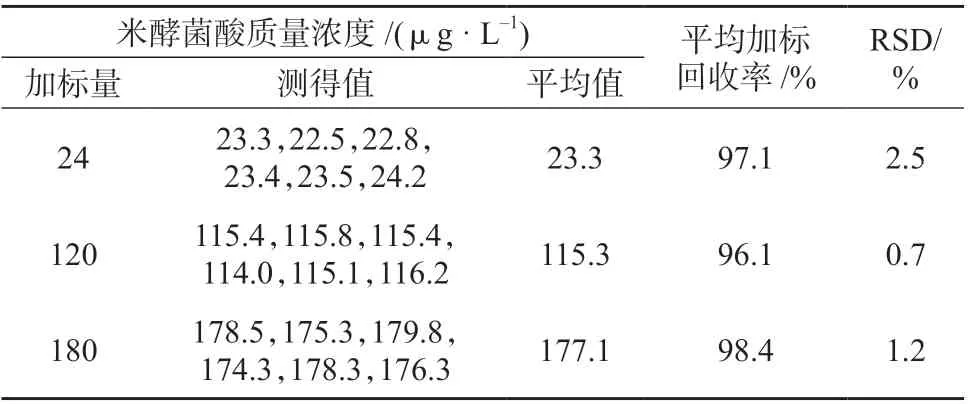
表3 加标回收试验结果
3 结语
建立了一种HPLC-MS/MS 方法快速检测米粉中米酵菌酸浓度。该方法对仪器条件和样品前处理过程进行了优化,具有检测快速、准确、检出限低、灵敏度高等特点,适用于米粉中米酵菌酸的检测。
猜你喜欢
当代水产(2022年4期)2022-06-05
中国药学药品知识仓库(2021年18期)2021-02-28
中国化工贸易·下旬刊(2019年10期)2019-10-21
农家科技(2019年1期)2019-03-13
分析化学(2017年12期)2017-12-25
食品界(2016年10期)2016-09-10
科技与创新(2016年7期)2016-04-20
科技与创新(2015年24期)2015-12-21
科技与创新(2015年22期)2015-12-02
中国信息化·学术版(2013年3期)2013-06-25
