
基于Au@N-CDs纳米复合材料电化学传感器检测左氧氟沙星
2022-09-30 08:27:26曹功勋刘翠娥郑凤英刘凤娇黄永俊骆嘉燚黄昭景李顺兴
分析科学学报 2022年4期
曹功勋, 刘翠娥, 满 珊, 郑凤英,2, 刘凤娇,2, 黄永俊,骆嘉燚, 黄昭景, 李顺兴*,2
(1.闽南师范大学化学化工与环境学院,闽南师范大学,福建漳州 363000;2.现代分离分析科学与技术福建省重点实验室&污染监测与控制福建省高校重点实验室,闽南师范大学,福建漳州 363000)
由于治疗细菌感染的广谱性,抗生素被过度使用,以致环境中其浓度水平不断升高[1],对生态系统和人类健康产生潜在危害,目前抗生素污染监测已成为全球关注热点[2]。我国是抗生素生产和使用大国,抗生素已成为我国水环境优先检出新兴污染物。据《中国抗菌药物管理和细菌耐药现状报告(2018)》,左氧氟沙星(Levofloxacin,LEV)在2017年抗菌药物消耗量(DDDs)排名前三位,是目前临床常用喹诺酮类抗菌药物。因此,开发有效、快捷、成本适宜的LEV检测法至关重要。目前LEV常用检测法有荧光光谱分析法[3]、核磁共振法[4]、液相色谱-质谱(LC-MS)法[5]、高效液相色谱(HPLC)法[6]等。但这些方法设备昂贵,且存在样品前处理复杂、成本高、分析时间长等不足,而电化学方法具有快速,便捷,成本低[7,8]等优点。
碳纳米材料作为新型催化剂或催化剂载体,具有优异导电、化学稳定性和大的比表面积等优势;Au纳米材料具有抗氧性和生物相容性等特点。由于金/碳纳米复合物具有两者双重特性,单分散掺杂石墨烯量子点包覆金纳米粒子兼具稳定和优异的生物相容性[9],Au纳米簇/碳量子点骨架复合材料表现出过氧化物酶活性和单激发/双重发射特性[10],亲水型Au@CDs纳米复合物可作为抗真菌剂[11],但基于Au/CDs纳米复合材料电催化活性和底物亲合性的传感器研究迄今未见报道。
本文利用氮掺杂碳点(N-CDs)的还原性原位合成Au纳米粒子(AuNPs),制备Au@N-CDs纳米复合材料,利用该材料对LEV的亲合选择性和电催化性能,定量测定LEV。
1 实验部分
1.1 主要仪器和试剂
Dimension Edge原子力显微镜(德国,布鲁克);JEM-2100F场发射透射电子显微镜(日本,JEOL公司);ESCALAB 250 Xi型X射线光电子能谱仪(美国,Thermo Fisher Scientific公司);Nicolet iS10傅立叶变换红外光谱仪(美国,Thermo Fisher Scientific公司);X射线粉末衍射仪(日本,Rigaku公司);SPECORD©200 PLUS型紫外-可见分光光度计(德国,耶拿分析仪器股份公司);Cary Eclipse荧光分光光度计(美国,Agilent公司);Nano ZS90 Zeta电位分析仪(英国,马尔文仪器有限公司);CHI 660E电化学工作站(上海辰华仪器有限公司)。
牛血清白蛋白(BSA)(≥98%,澳大利亚);NaOH、H3PO4、H3BO3、KCl、一水合柠檬酸、无水Na2HPO4、KH2PO4均购买于西格玛奥德里奇(上海)贸易有限公司;HAc(≥98%,色谱纯)、利福平、罗红霉素、左氧氟沙星、红霉素均购买于上海阿拉丁生化科技股份有限公司;实验用水为美国Millipore超纯水仪器制备的超纯水(18.2 MΩ·cm)。
1.2 Au@N-CDs的制备
参照文献报道[12],对N-CDs合成方法稍作修改:将0.0300 g BSA溶于15 mL超纯水,快速混合,加入15 mL无水乙醇,冰水浴中超声30 min,转至50 mL聚四氟乙烯内衬中,于高压釜中180 ℃恒温加热12 h,冷却至室温,0.22 μm滤膜过滤后,转入3 500 D透析袋透析48 h,冷冻干燥,得固体粉末N-CDs。
将5.0 mg N-CDs超声溶于Britton-Robinson(B-R)缓冲溶液(pH=4.0,100.0 mL),逐滴加入HAuCl4(12 mmol/L,2.0 mL)溶液,将混合溶液在37 ℃水浴恒温振荡器上孵育12 h,再置于4 ℃环境中24 h,11 000 r/min离心20 min,经超纯水洗涤3次,冷冻干燥,得黑色粉末Au@N-CDs,置于4 ℃环境中保存备用。制备流程见图1。
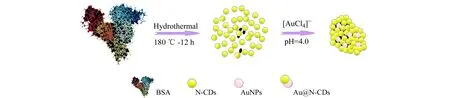
图1 制备Au@N-CDs的流程图Fig.1 Preparation of Au@N-CDs with water bath incubation
1.3 Au@N-CDs修饰电极制备
N-CDs和Au@CDs修饰液:将1.5 mg的N-CDs或Au@N-CDs超声分散于0.05% Nafion溶液中,分别得到1.5 mg/mL的浅褐色N-CDs修饰液和黑色Au@N-CDs修饰液。依次用超纯水、无水乙醇和超纯水超声清洗用麂皮抛光的玻碳电极(GCE),用高纯氮气吹干,分别滴涂10 μL修饰液,于常温下自然干燥,分别制得N-CDs修饰电极(N-CDs/GCE)和Au@N-CDs修饰电极(Au@N-CDs/GCE)。
1.4 左氧氟沙星的检测
在室温下,分别以N-CDs/GCE(或Au@N-CDs/GCE)、Ag/AgCl电极、铂丝电极为工作、参比和辅助电极,于含有不同浓度LEV的磷酸盐缓冲液(PBS)或人体尿液(pH=6.0)样品中,在电位0.4 V作用下,富集125 s,在0.6~1.3 V电位范围内,通过微分脉冲伏安法绘制工作曲线。
2 结果与讨论
2.1 Au@N-CDs制备及其形貌与组成
在N-CDs(0.05 mg/mL,5 mL)的不同pH值B-R缓冲溶液中,添加HAuCl4溶液(20 mmol/L,50 μL),用快速混匀器快速振荡,于37 ℃孵化12 h,置于4 ℃条件下陈化24 h,当pH=4.0时,反应液底部有一定量的黑色沉淀生成。由于N-CDs等电点在pH=4.0附近,在等电点时,颗粒净电荷为零,没有相同电荷的排斥作用,分子间作用力弱,颗粒易碰撞而凝聚沉淀。N-CDs为浅褐色,而沉淀为黑色,对照组未生成黑色沉淀,这表明N-CDs和HAuCl4发生反应。N-CDs可作为还原剂原位还原HAuCl4,并在合成过程中吸附包裹生成的AuNPs,从而形成Au@N-CDs。
在pH=4.0,含N-CDs(0.05 mg/mL,5 mL)的B-R缓冲溶液中,添加不同浓度HAuCl4(0、6、8、10、12、14、16、18、20 mmol/L,100 μL)溶液,制备Au@N-CDs,通过紫外-可见和荧光发射光谱测试制备效果。紫外吸收在520~560 nm处均出现Au吸收峰,吸光强度先增后减,这是由于HAuCl4浓度过高,初生AuNPs较多,N-CDs不能及时还原,体系高比表面能使得AuNPs团聚,因此在12 mmol/L的HAuCl4浓度时,吸光值最大(图2(a)),此时生成的AuNPs浓度最高,而N-CDs由于同AuNPs结合引起N-CDs荧光猝灭(图2(b))。
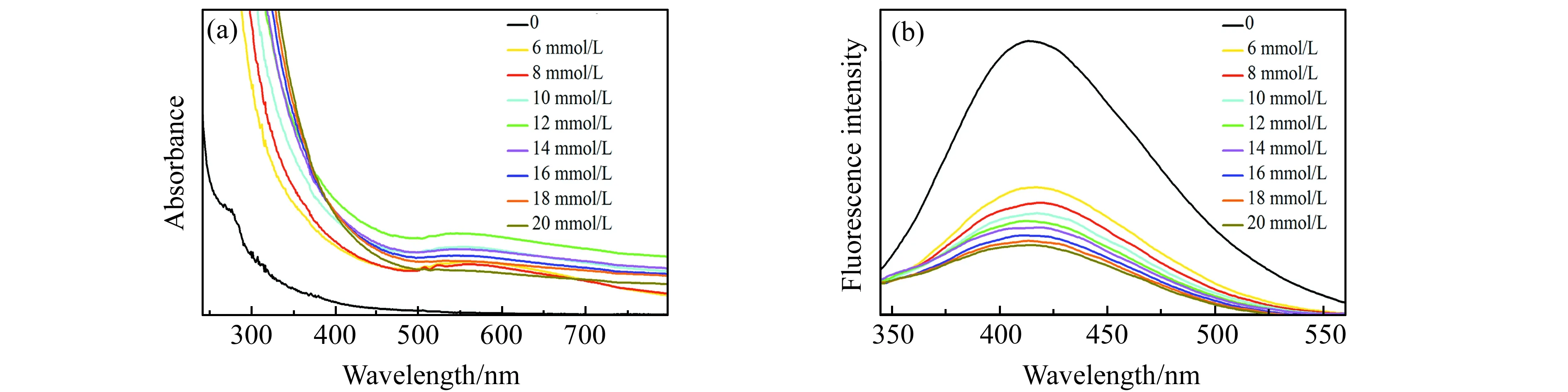
图2 (a)HAuCl4不同浓度下Au@N-CDs紫外-可见吸收光谱;(b)Au@N-CDs的荧光发射光谱Fig.2 (a) Influence of HAuCl4 concentrations on ultraviolet-visible absorption spectra;(b) Fluorescence emission spectra of Au@N-CDs
由高分辨透射电镜(HR TEM)图3(a)可知,荧光黄椭圆圈内图像具有0.20 nm晶格条纹间距与Au(200)晶面相吻合[13,14];晶格条纹间距0.32 nm与层间石墨烯(002)晶面相一致[15,16],这表明Au3+已被N-CDs还原。XRD(图3(b))图谱同检索JCPDS no.1-1172对比,宽衍射峰2θ=23.16°对应碳的石墨化结构[17,18],2θ=38.04°、44.24°、64.53°、77.50°均为Au特征衍射峰[9],分别对应晶面(111)、(200)、(220)、(311)[19]。上述结果证明成功合成Au@N-CDs。原子力显微镜(AFM)图及3D模式图3(c)表明,Au@N-CDs厚度约2 nm,AuNPs和N-CDs组装在二维平面,第三维度超薄[20],便于负载于GCE。元素映射分析图4中,材料由Au、C和N组成,N和C分布密实,Au分散于C和N中,即AuNPs嵌套在N-CDs基质中,构成Au@N-CDs。
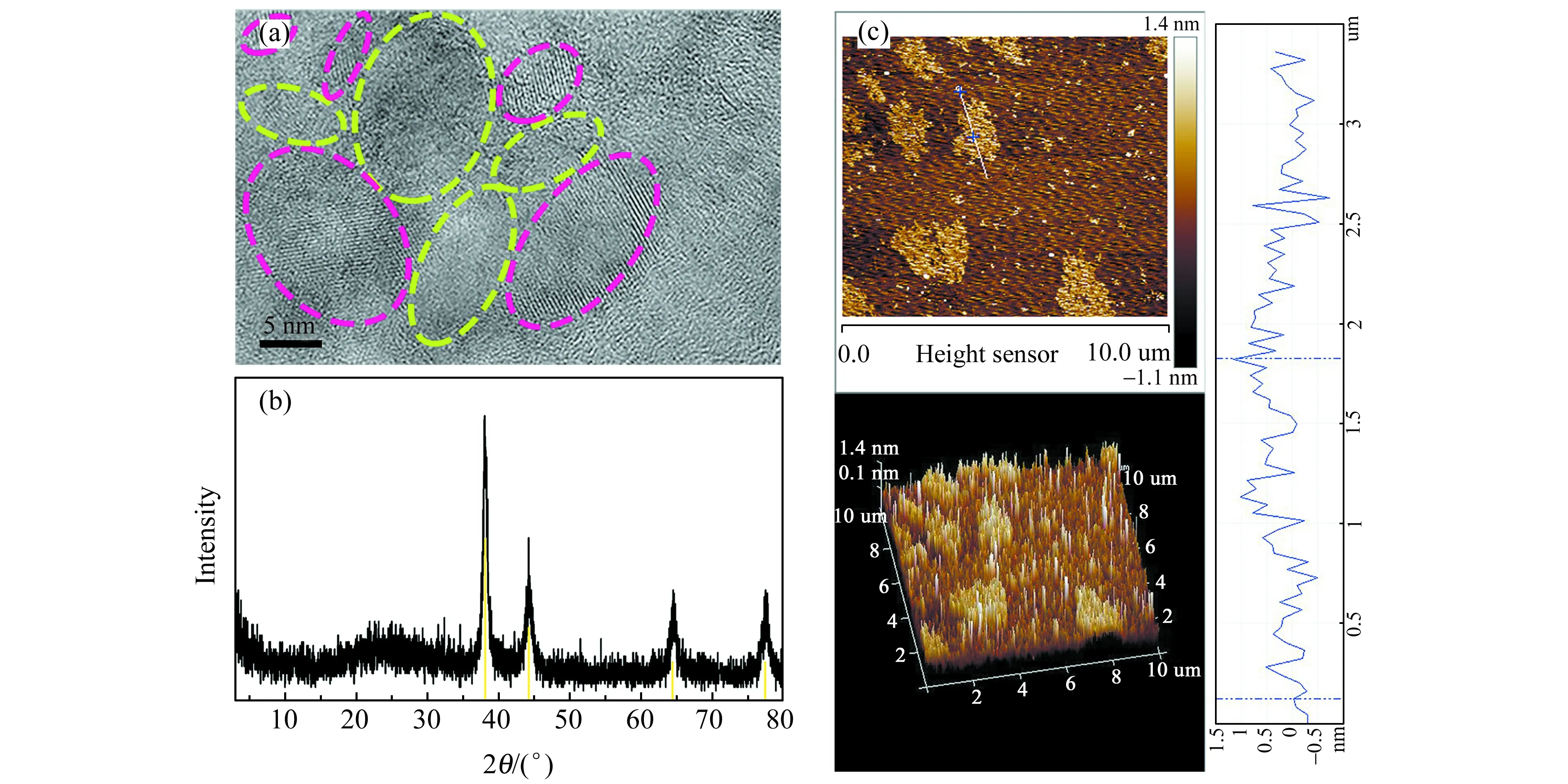
图3 (a)Au@N-CDs的透射电镜(TEM)图(插图为HRTEM图);(b) X射线衍射图;(c)原子力显微镜图和相应粒子高度分布图Fig.3 (a) HR TEM image of Au@N-CDs;(b) X-ray diffraction pattern of Au@N-CDs;(c) Atomic force microscope pattern and corresponding particle height distribution pattern of Au@N-CDs

图4 (a)Au@N-CDs的C、N、Au元素映射分析叠加图;(b、c、d)C、N元素和Au元素的映射分析图Fig.4 (a)Overlay of C,N,Au element mapping analysis of Au@N-CDs;(b,c,d)Mapping analysis diagram of C,N and Au
傅立叶红外(FTIR)光谱测试N-CDs和Au@N-CDs官能团,结果如图5所示。N-CDs在3 290 cm-1、2 962 cm-1、1 664 cm-1和1 101 cm-1处有显著的吸收峰,这与N-H[21],C-H[22],C=O[23,24]和C-O[25,26]振动相关;Au@N-CDs相应红外吸收峰发生移动,N-H、C-H、C=O、C-O分别移至3 421 cm-1、2 925 cm-1、1 647 cm-1和1 084 cm-1,说明N-CDs和AuNPs间有交互作用。

图5 Au@N-CDs和N-CDs的傅立叶变换红外光谱图Fig.5 Fourier transform infrared spectra of Au@N-CDs and N-CDs
X射线光电子能谱(XPS)表征Au@N-CDs全扫描图谱表明,样品由C、N、O、S和Au组成,元素含量分别为C 62.01%、N 12.4%、O 22.32%、S 0.63%及Au 2.64%。XPS高分辨C 1s谱(图6(a))中3个峰结合能分别为283.93 eV、285.03 eV和286.88 eV,归于C=C/C-C、C-C/C-S/C-N和C=O[17,18]。N 1s谱(图6(b))在399.38 eV和401.33 eV处的峰归于C-N、N-H[27]。O 1s谱(图6(c))显示两个相对氧态,COO-(530.98 eV)、C-OH或C-O-C(532.38 eV)[28]。Au 4f谱(图6(d))分别在84.23eV和87.88eV处的两峰为Au04f7/2和Au04f5/2的光电子作用[16]。
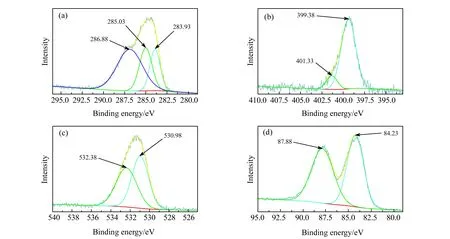
图6 Au@N-CDs的X射线光电子能谱(XPS)表征Fig.6 XPS of Au@N-CDs(a) C 1s spectrum;(b) N 1s spectrum;(c) O 1s spectrum;(d) Au 4f spectrum.
2.2 Au@N-CDs/GCE的电化学表征
在含0.1×10-3mol/L KCl的5.0×10-3mol/L [Fe(CN)6]3-/4-溶液中,电化学阻抗谱(EIS)测试结果如图7(a)所示。结果表明,裸GCE、N-CDs/GCE、Au@N-CDs/GCE的EIS中R2的值分别为118.7 Ω、4 023 Ω和15 270 Ω,电荷转移效率越来越低,同时低频区斜线倾斜率减小,电极离子导电率越来越小。在[Fe(CN)6]3-/4-溶液中测试GCE、N-CDs/GCE和Au@N-CDs/GCE的循环伏安图,得图7(b)。GCE、N-CDs/GCE和Au@N-CDs/GCE的氧化还原峰电流依次减小,N-CDs和Au@N-CDs在GCE上修饰,降低了电极的电子转移速率,这与EIS分析结果一致,表明材料成功修饰在电极上。
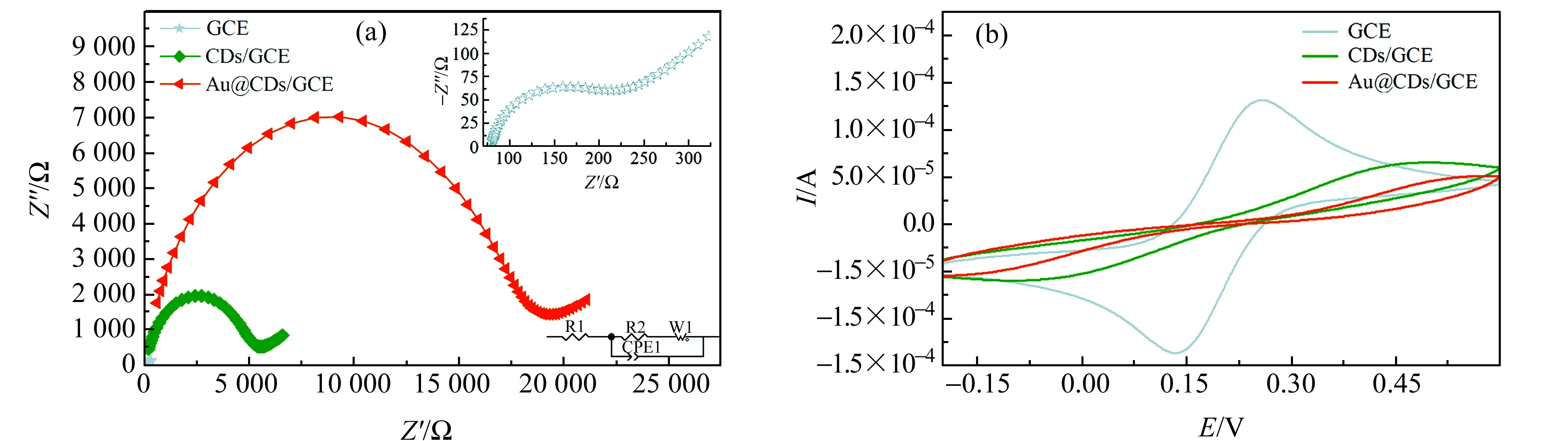
图7 (a)GCE、N-CDs /GCE和Au@ N-CDs /GCE的电化学阻抗谱(EIS);(b)GCE、N-CDs/GCE和Au@N-CDs/GCE的循环伏安图Fig.7 (a) EIS of GCE,N-CDs/GCE and Au@N-CDs/GCE; (b) Cyclic voltammograms of GCE,N-CDs/GCE and Au@N-CDs/GCE
2.3 Au@N-CDs/GCE测定LEV条件及性能
在0.01 mol/L的PBS(pH=6.0)测试体系中,以循环伏安法检测Au@N-CDs/GCE在10~300 mV/s扫描速率范围内对LEV的电化学响应,将不同扫描速率的CV曲线叠加,得图8(a)。LEV氧化峰电流随CV扫描速率增加而增大,Ip与扫速呈良好线性关系,如图8(b),其线性回归方程为:Ip=3.969×10-7v+2.078×10-5(R=0.9935),表明LEV在Au@N-CDs/GCE上的反应为表面吸附控制的电极反应过程。

图8 (a)不同扫速下1 mmol/L LEV在Au@CDs/GCE上的循环伏安图;(b)不同循环伏安扫描速率和LEV氧化峰电流Ip之间的关系图Fig.8 (a) Cyclic voltammogram of LEV(1 mmol/L) on Au@N-CDs/GCE under different scanning rate;(b) Relationship between different cyclic voltammetry scan rates and LEV oxidation peak current Ipscan rate:10,30,60,90,120,150,180,210,240,270,300 mV/s.
在0.1~0.8 V富集电位范围内,随电位值增大,LEV的氧化峰电流Ip先增后减。当富集电位为0.4 V时,Ip最大(图9(a))。随富集时间增加,Ip增大,但当富集时间超过125 s,Ip增幅微小且趋于稳定(图9(b)),可见电极对LEV的表面吸附富集已达饱和,因此选择富集时间为125 s。
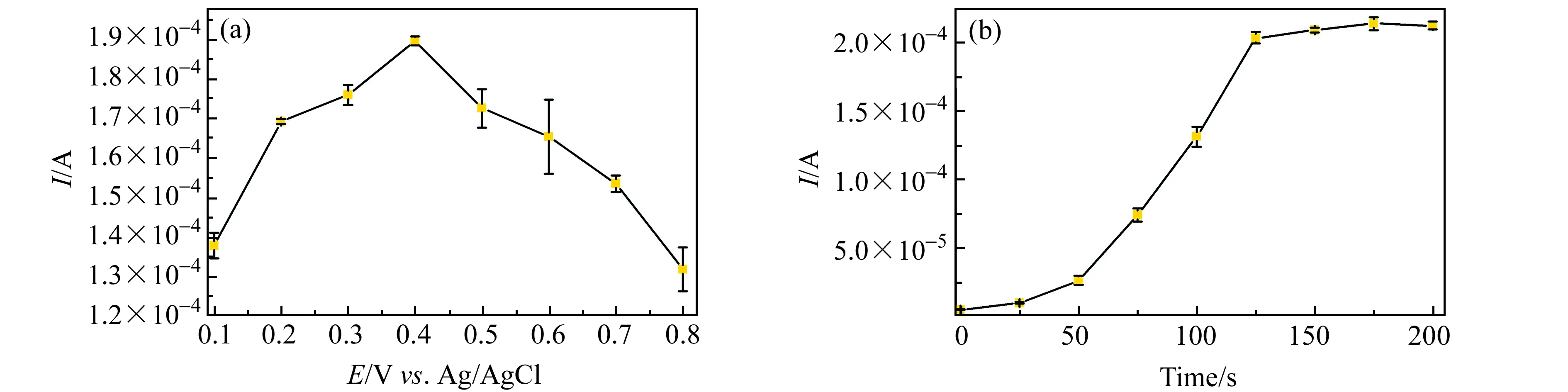
图9 (a)富集电位、(b)富集时间对Au@N-CDs/GCE测定LEV的影响Fig.9 (a)Influence of enrichment potential and (b)enrichment time on the determination of LEV with Au@N-CDs/GCE
当pH值从4.0增至7.0,电流Ip呈现先增后减(图10(a)),在pH=6.0时Ip达到最高值。此外,Ep随pH值增大而发生电位负移,与石墨烯/金纳米复合材料修饰GCE结果相似[29],这主要是电极反应过程有氢离子的参与因此发生移动,其具体反应机理如图10(b)所示。

图10 (a)不同pH的PBS中LEV在Au@N-CDs/GCE上的循环伏安图;(b)检测LEV的反应机理图Fig.10 (a)Cyclic voltammogram of LEV on Au@N-CDs/GCE in phosphate buffer solution with different pH;(b)Reaction mechanism for detecting LEV
以Au@N-CDs/GCE为工作电极,以差分脉冲伏安法(DPV)测定LEV,结果如图11(a)和11(b)。在2.5×10-6~1.0×10-4mol/L内,LEV的Ip值与浓度c呈良好的线性关系,线性回归方程为:Ip(μA)=0.08455c(μmol/L)+4.102(R=0.9907),检出限(3σ/k)为1.0×10-6mol/L。
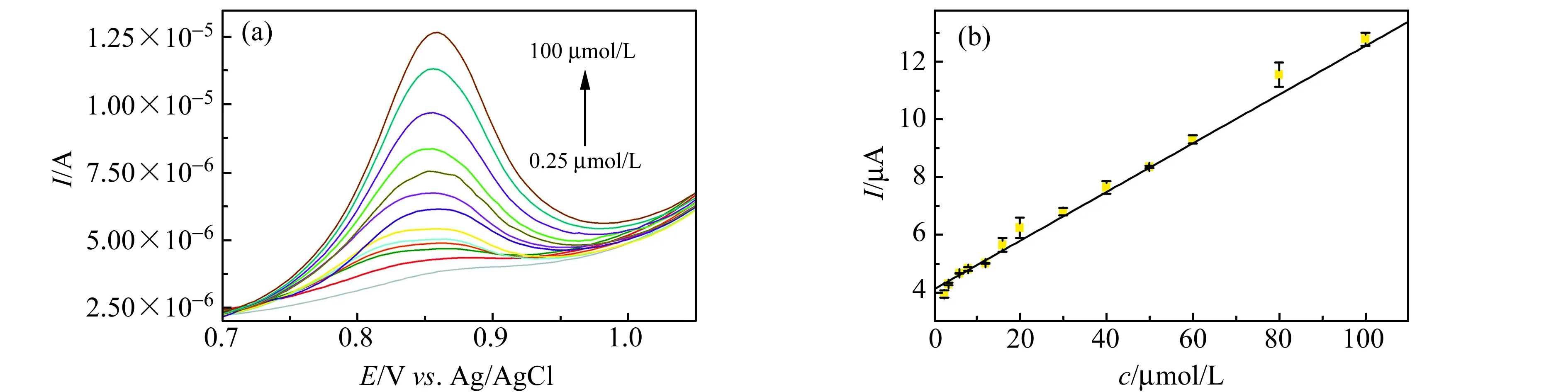
图11 (a) Au@N-CDs/GCE测定2.5、3.5、6.0、8.0、10、16、20、30、40、60、80、100 μmol/L LEV的差分脉冲伏安法图;(b)不同LEV浓度和LEV氧化峰电流Ip之间的关系图Fig.11 (a) Differential pulse voltammogram of LEV(0.25 - 100 μmol/L) on Au@N-CDs/GCE;(b)Relationship between concentrations and oxidation peak current Ip for LEV
同样制备6根电极考察Au@N-CDs/GCE对LEV检测的重现性,测定1.0×10-5mol/L的LEV,相对标准偏差(RSD)为2.98%,因此Au@N-CDs/GCE对LEV检测有较好的重现性。
同时也研究了其他抗生素作为干扰物质对于LEV电化学信号的影响。先检测LEV浓度为5.0×10-5mol/L的氧化峰电流,后再向LEV溶液中加入相同含量的利福平、罗红霉素、红霉素。上述物质对LEV电化学行为无明显影响,这表明Au@CDs/GCE对检测LEV具有一定的抗干扰能力。这可能是利福平电化学检测氧化还原电位差较大[30],因此利福平不干扰左氧氟沙星的电化学信号。罗红霉素、红霉素因其无-COOH,故其吸附力较小,对LEV干扰也较小。
2.4 人体尿液样品分析
LEV在血浆和尿液中的立体化学结构稳定,人体对LEV的代谢量很低,主要从尿中排出。因此,对稀释处理后购置的高浓度质控人体尿液采用标准加入法,测试Au@N-CDs/GCE对LEV电化学检测的实际应用能力,计算LEV的回收率和相对标准偏差(RSD)。实验结果如表1所示,所获得的样品回收率为93.0%~107.4%,且RSD在6.8%之内,因此Au@N-CDs/GCE具有应用于人体尿样和废水中LEV测定的可行性。

表1 样品中LEV的检测结果
3 结论
本研究制备的Au@N-CDs纳米复合材料,在pH=6.0的PBS中测定LEV的含量,在电位0.4 V下富集125 s后测试效果较好,Au@N-CDs/GCE检测LEV线性范围为2.5×10-6~1.0×10-4mol/L,检出限为1.0×10-6mol/L。Au@N-CDs/GCE检测LEV同时,对利福平,罗红霉素、红霉素有抗干扰作用,对人工尿液采用标准加入法,证明了Au@N-CDs/GCE在尿液和废水中LEV检测的可行性。
猜你喜欢
中南民族大学学报(自然科学版)(2022年3期)2022-05-08 03:51:04
节能与环保(2022年3期)2022-04-26 14:32:44
食品安全导刊(2021年20期)2021-11-28 00:56:56
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19 09:03:04
表面工程与再制造(2019年6期)2019-08-24 06:40:08
资源节约与环保(2018年1期)2018-02-08 02:18:27
池州学院学报(2017年3期)2017-10-16 01:38:37
电镀与环保(2016年2期)2017-01-20 08:15:26
现代工业经济和信息化(2016年12期)2016-05-17 05:37:52
中国有色冶金(2014年6期)2014-08-10 12:29:04
