
超高效液相色谱-串联质谱法测定叔胺类药品中硫酸二甲酯基因毒性杂质
2022-09-26 07:02:08巩丽萍杭宝建咸瑞卿杨明正张迅杰
色谱 2022年9期
巩丽萍, 杭宝建, 咸瑞卿, 杨明正, 张迅杰, 魏 霞
(山东省食品药品检验研究院, 国家药品监督管理局仿制药研究与评价重点实验室,山东省仿制药一致性评价工程技术研究中心, 山东 济南 250101)
硫酸二甲酯是重要的化工原料之一[1,2],在有机合成中作为甲基化试剂广泛用于制造二甲基亚砜、咖啡因、可待因、香草醛、安替比林、氨基比林、甲氧苄氨嘧啶等药物以及农药乙酰甲胺磷等,有些药物在生产过程中也会产生硫酸二甲酯[3]。硫酸二甲酯为剧毒品,具有遗传毒性和致癌性,且具有强烈的腐蚀性,能在动物体内组织中缓慢水解生成甲醇和硫酸,其毒性是由未分解的分子和水解后的生成物共同作用产生。药物中残留的硫酸二甲酯在过量(《美国药典》《欧洲药典》规定原料药中硫酸二甲酯的含量不得过0.000 038%)时会对人体产生巨大的危害,因此非常有必要控制药物中硫酸二甲酯的残留量[4,5]。
目前对硫酸二甲酯的检测方法主要有气相色谱法、气相色谱-质谱联用法和液相色谱法[6-15],但准确测定药物中残留的硫酸二甲酯含量极具挑战性,因为硫酸二甲酯极性很大,不具备敏感的紫外吸收基团,因此传统方法在专一性、灵敏度、方法精密度和重复性等方面都存在很大缺陷,且检测过程中对溶剂的要求很严格,如溶液配制不允许使用水,必须使用纯度极高的乙腈,气相色谱-质谱联用法得到的谱图在低分子端会出现大量的碎片峰,干扰检测的精准度。
本研究以氨基比林作为衍生试剂,采用超高效液相色谱-串联质谱法建立了氨基比林、咖啡因、替加氟等叔胺类药物中硫酸二甲酯的检测方法。硫酸二甲酯具有甲氧基基团,可以和氨基比林发生N-烃基化反应生成甲基化氨基比林(季铵盐),样品前处理简便,生成的甲基化氨基比林稳定性高、干扰小、质谱离子化好、响应高,方法快速、准确、灵敏,可用于上述叔胺类药品的质量控制。
1 实验部分
1.1 仪器与试药
AB SCIEX Triple Quad 6500+超高效液相色谱-三重四极杆质谱联用仪(AB公司;美国); XSE205电子天平(Metter Toledo公司,瑞士); KQ-300GDV型恒温数控超声器(昆山市超声仪器有限公司); Vortex-6涡旋振荡器(海门市其林贝尔仪器制造有限公司)。甲醇、丙酮(色谱纯,美国赛默飞公司),甲酸(色谱纯,美国ACS公司),乙酸铵(色谱纯,国药集团化学试剂有限公司),去离子水(18.2 MΩ·cm,由美国Merck-Millipore公司生产的Millipore Milli-Q Advantage超纯水系统制得)。对照品:硫酸二甲酯(山东西亚化学公司),纯度99.5%。样品:氨基比林、咖啡因、替加氟原料药共9批。
1.2 色谱、质谱条件
液相色谱条件:色谱柱为Waters Atlantis HILIC C18色谱柱(100 mm×2.1 mm, 3.0 μm),以10 mmol/L乙酸铵水溶液-0.1%甲酸甲醇溶液(50∶50, v/v)为流动相进行等度洗脱,流速0.3 mL/min,柱温40 ℃,进样量1 μL。
质谱条件:电喷雾电离(ESI)源,正离子扫描模式,多反应监测;离子源温度:550 ℃。定性、定量离子对、锥孔电压、碰撞能量见表1。

表 1 甲基化氨基比林的质谱采集离子信息Table 1 Ion information of methylated aminopyrine acquired by MS
1.3 衍生剂、空白溶液的制备
取氨基比林1.000 g,精密称定,置于100 mL量瓶中,加水溶解并稀释至刻度,摇匀,作为衍生剂溶液;取0.5 mL水,加衍生剂溶液0.5 mL, 40 ℃反应2 h,作为空白溶液。
1.4 对照品溶液的制备
取硫酸二甲酯对照品适量,精密称定,加丙酮溶解并定量稀释,制成20 μg/mL的对照品储备溶液。精密量取溶液0.05 mL,加水稀释制成4 ng/mL的对照品工作溶液(临用新制);精密量取溶液0.5 mL,精密加入衍生剂溶液0.5 mL,摇匀,40 ℃反应2 h,作为对照品溶液。
1.5 供试品溶液的制备
氨基比林原料药:取本品适量,精密称定,加水溶解并定量稀释,制成5 mg/mL的溶液;取溶液1.0 mL,于40 ℃放置2 h,作为氨基比林供试品溶液。
替加氟等叔胺类原料药:取本品适量,精密称定,加水溶解并定量稀释,制成5 mg/mL的溶液;精密量取溶液0.5 mL,精密加入衍生剂溶液0.5 mL,摇匀,40 ℃反应2 h,作为其他叔胺类供试品溶液。
2 结果与讨论
2.1 反应产物的确认及反应温度的考察
硫酸二甲酯是甲基化试剂,和氨基比林发生N-烃基化反应生成季铵盐(见图1)。反应过程中温度是影响反应效率的主要因素,因此对不同反应温度进行了考察。
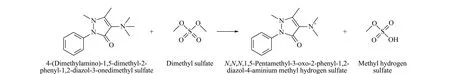
图 1 硫酸二甲酯与氨基比林的反应式Fig. 1 Reaction formula of dimethyl sulfate and aminopyrine
考察实验过程如下:精密称定硫酸二甲酯(相对分子质量为126.13)适量,加丙酮溶解并稀释制成浓度为4.5 mmol/L的溶液。精密称定氨基比林(相对分子质量为231.29)适量,加水溶解并稀释制成浓度为45.0 mmol/L的溶液。取上述硫酸二甲酯溶液和氨基比林溶液各1 mL,混匀,分别于30、40、50 ℃反应2 h,取上述反应液各1.0 mL,分别置于100 mL量瓶中,用水稀释至刻度,摇匀,作为考察样品溶液。同法制备空白溶液。采用Thermo QE Plus高分辨质谱联用仪进行分析,计算出不同温度下的反应效率,并对反应产物进行定性确认。
考察样品溶液和空白溶液的一级高分辨离子色谱图和紫外光谱图分别见图2a和图2b。考察样品溶液中因氨基比林在反应体系中过量,故有剩余没反应的氨基比林,因此会采集到衍生产物甲基化氨基比林和剩余反应物氨基比林的质谱信息,同时采集了剩余反应物氨基比林的紫外光谱;空白溶液中只有氨基比林,因此一级高分辨离子色谱图只有氨基比林的信息。由于采用的是高分辨质谱,精度高,可以对反应产物进行定性确认,由一级高分辨离子色谱图可以看出,氨基比林与硫酸二甲酯的反应液中检测到反应产物的精确质量数为246.124 6,与氨基比林甲基化产物的理论精确质量数246.124 6一致,确认了反应产物为甲基化氨基比林。以氨基比林在考察样品溶液和空白溶液中233 nm测定的紫外光谱图峰面积差值计算反应效率。随着温度的升高,硫酸二甲酯和氨基比林的反应效率增加,在40 ℃时基本上达到完全反应,因此选择40 ℃作为衍生温度。

图 2 反应物、反应产物在(a)考察样品溶液和(b)空白溶液中的提取离子色谱图和紫外色谱图Fig. 2 Extraction ion and UV chromatograms of reactants and reaction products in (a) test sample and (b) blank solutions The reaction products were investigated by ultra-high performance liquid chromatography-electrostatic field orbitrap high resolution mass spectrometry (UHPLC-Orbitrap HRMS). The separation was performed on a Thermo Hypersil GOLD column (100 mm×2.1 mm, 3 μm) using 10 mmol/L ammonium acetate solution-0.1% formic acid methanol solution (50∶50, v/v) at a flow rate of 0.3 mL/min. The column temperature was set at 40 ℃, detection wavelength was 233 nm and injection volume was 1 μL.
2.2 氨基比林衍生剂的选择
吡啶是硫酸二甲酯发生甲基化反应的常用底物,是一个含有氮杂原子的六元杂环化合物,具有芳香性,而本研究发现的氨基比林为含有α,β不饱和酮结构的脂肪胺,吡啶环上的N原子相比氨基比林上的N原子因电子云轨道杂化、空间位置不同而活性较低,且硫酸二甲酯与胺的反应属于双分子亲核取代(SN2)反应,在SN2反应中,氨基比林表现出更强的亲核性,反应能够快速进行。与吡啶相比,氨基比林与硫酸二甲酯反应不需要高温,较低温度有利于硫酸二甲酯的稳定。吡啶与硫酸二甲酯反应后产物具有季铵盐的特性,但生成的季铵盐不利于其芳香性的保持,不够稳定,但是氨基比林生成的脂肪胺季铵盐稳定性好,因此本研究选取的氨基比林更适合作为甲基化试剂用于硫酸二甲酯的检测。
2.3 专属性
精密量取空白溶液、对照品溶液、供试品溶液各1 μL,分别进样测定。空白溶液与供试品溶液中其他杂质峰均不干扰硫酸二甲酯衍生后的甲基化氨基比林的检测,空白溶液中甲基化氨基比林保留时间处有峰值很小的本底色谱峰(见图2)。连续将空白溶液进样7次,计算该色谱峰峰面积的RSD值为6.17%,表明该色谱峰的稳定性良好,不影响硫酸二甲酯的检测。
2.4 标准曲线
分别精密量取对照品储备溶液(20 μg/mL)0.5、1.0、2.0、3.0、4.0 mL,分别置于100 mL量瓶中,加水稀释至刻度,摇匀。分别精密量取上述溶液0.5 mL,再分别精密加入0.5 mL衍生剂溶液,混匀,40 ℃反应2 h,进样测定。以甲基化氨基比林的峰面积为纵坐标(y),各溶液的质量浓度为横坐标(x, ng/mL),绘制标准曲线,结果表明在0.993 5~7.984 0 ng/mL范围内,线性关系良好,回归方程为y=17 901x-3 146,相关系数(r)为0.999 7。
2.5 检出限和定量限
基于空白溶液中存在本底峰,检出限和定量限采用《中国药典》2020年版四部通则9101中“基于响应值标准偏差和标准曲线斜率法”测定。取空白溶液连续进样7次,计算检出限为0.50 ng/mL(0.05 ppm),定量限为1.15 ng/mL(0.115 ppm)。
2.6 精密度
取对照品溶液连续进样6次,甲基化氨基比林峰面积的RSD值为2.80%,表明该方法的仪器精密度良好。
2.7 稳定性
取对照品反应溶液分别在0、2、4、6、20 h进样测定,甲基化氨基比林峰面积的RSD值为4.32%,表明反应产物稳定性良好。
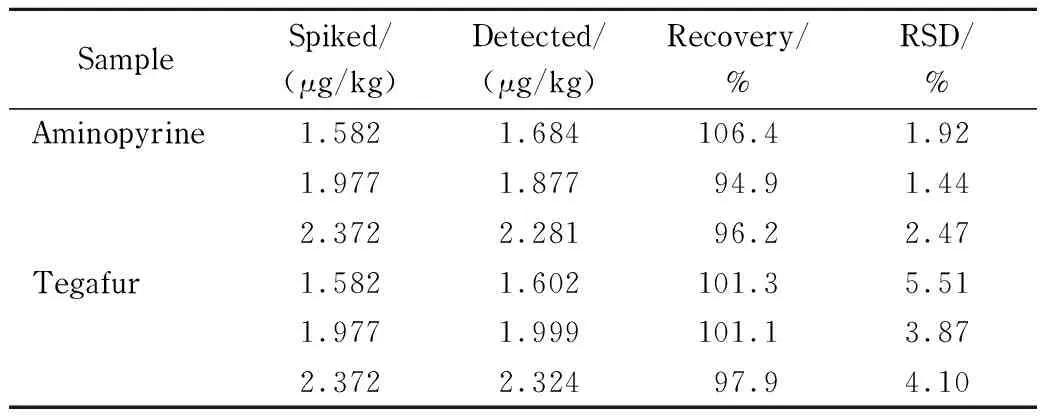
表 2 三个水平下硫酸二甲酯的加标回收率(n=3)Table 2 Spiked recoveries of dimethyl sulfate at three levels (n=3)
2.8 回收率
本方法通过向阴性氨基比林、替加氟样品中分别添加低、中、高3个水平的硫酸二甲酯对照品溶液,每个水平进行3次平行试验,计算方法回收率,结果见表2。硫酸二甲酯的平均回收率为94.9%~106.4%,相对标准偏差为1.44%~5.51%,满足实际样品的检测需求。
2.9 样品测定
取氨基比林、咖啡因、替加氟共9批原料药按1.5节制备供试品溶液,进样测定。按外标法计算硫酸二甲酯的含量,9批样品中均未检出该杂质,表明氨基比林、咖啡因、替加氟原料药生产企业在生产工艺对硫酸二甲酯基因毒杂质控制良好。
3 结论
本文建立了超高效液相色谱-串联质谱测定氨基比林等叔胺类药品中硫酸二甲酯基因毒杂质含量的检测方法,该方法将氨基比林作为衍生剂,与药品中残留的硫酸二甲酯发生反应生成甲基化氨基比林,通过检测甲基化氨基比林间接测得药品中硫酸二甲酯的含量,进行了方法学验证,并将方法用于实际样品的测定。与现有技术相比,该方法离子峰峰形好,无其他碎片杂峰干扰,方法灵敏度高、精密度和重复性好,结果可靠,可用于氨基比林等药品中硫酸二甲酯基因毒杂质的测定。本研究有助于药品生产企业对生产工艺中基因毒杂质的控制,同时为药品监管提供技术支撑。
猜你喜欢
云南化工(2021年10期)2021-12-21 07:33:42
现代畜牧科技(2021年9期)2021-10-13 06:38:40
精细石油化工(2015年3期)2015-12-14 09:07:42
河南科技(2015年2期)2015-02-27 14:20:35
同位素(2014年2期)2014-04-16 04:57:13
济源职业技术学院学报(2014年3期)2014-02-28 02:35:42
江西理工大学学报(2013年1期)2013-03-20 14:57:07
郑州大学学报(理学版)(2013年3期)2013-03-11 20:30:40
食品科学(2013年24期)2013-03-11 18:30:37
哈尔滨医药(2012年4期)2012-12-28 09:03:48
