
受阻胺类化合物1-环己氧基-2,2,6,6-四甲基哌啶-4-酮的合成新方法
2022-09-26 07:35:16张秀芹何燕岭梁顺全
化工技术与开发 2022年9期
张秀芹,何燕岭,梁顺全
(中山苏特宝新材料有限公司,广东 中山 528441)
在聚烯烃高分子材料中,相比紫外吸收剂和光屏蔽剂等,作为近年来发展最快的新型添加剂,受阻胺光稳定剂[1]的光稳定效果优异[2],因此得到了大量应用。近年来的研究发现,受阻胺类化合物同样具有高效的阻燃性能,其在受热燃烧时,会分解形成一系列的自由基如OR、哌啶氮、R、NO等,可以有效减少高分子材料燃烧过程中自由基的产生,从而实现阻燃性能[3]。
图1为受阻胺的化学式,可以看出,受阻胺为含氮六元杂环化合物,具有立体效应,其中R为取代基,可以是H氢、R烷基、OR烷氧基等;A为辅助基团,与哌啶基连接。

图1 受阻胺的化学结构式Fig.1 The Chemical formula of obstructed amines
实验结果证明[4],由于取代基的差异,受阻胺化合物的碱性强弱也存在差异,碱性由强到弱依次为N-H、N-R、N-OR。从研究成果可知,碱性最弱的N-OR为阻燃受阻胺[5],其余的均不具备阻燃特性。
国内的学者对受阻胺类阻燃剂进行了不少研究,大多都是以TMP(2,2,6,6-四甲基哌啶酮)为起始原料,通过催化反应获得氮氧自由基中间产物,再在不同的氧化还原引发体系中,在催化剂的作用下发生相转移,中间产物与环己烷发生合成反应,获得1-环己氧基-4-氧-2,2,6,6-四甲基哌啶酮,各反应的差异在于氧化剂与还原剂。陈苗琴等人[6]以过氧化氢为氧化剂,还原剂为FeSO4·7H2O,制备了1-环己氧基-4-氧-2,2,6,6-四甲基哌啶酮。朱新斌等人[7]以叔丁基过氧化氢为氧化剂,溴化铜为还原剂,制备了1-环己氧基-4-氧-2,2,6,6-四甲基哌啶酮。有少量国外的专利公开了1-环己氧基-4-氧-2,2,6,6-四甲基哌啶酮的合成研究成果。Galbo等人[8]在Fe2+化合物与有机酸的催化作用下,阻聚剂701与C6H12发生环氧基取代反应,获得了1-环己氧基-4-氧-2,2,6,6-四甲基哌啶酮,此方法存在转化率不高、工艺条件要求高等缺点。Basbas等人[9]在Cu+化合物的催化作用下,2,2,6,6-四甲基哌啶氮氧自由基与C7H12O发生反应获得了中间体,但此方法的反应速率低,无法实现工业化生产。Dichtl等人[10]建立了1-环己氧基-4-氧-2,2,6,6-四甲基哌啶酮的简单可靠的合成方法,使用过氧化物、Cu+化合物等为催化剂,阻聚剂702与酮类化合物发生取代反应,制备了1-环己氧基-4-氧-2,2,6,6-四甲基哌啶酮。尽管此方法有一定的优势,但由于选择性较低,目标产物相对较少,产率偏低,因此无法应用于工业生产中。Kai-Uwe等人[11]在CuCl的催化作用下,阻聚剂701与C7H12O发生反应获得了目标产物,但C7H12O的价格昂贵,生产成本高,不利于规模化生产。
本方法以t-BuOOH为氧化剂,FeSO4·7H2O为还原剂构成引发体系,以阻聚剂4-羟基-2,2,6,6-四甲基哌啶氮氧自由基(阻聚剂701)和环己烷为原料,通过一步反应获得目标产物,研究了反应条件和反应机理,并对合成产物进行了表征与纯度分析。
1 实验部分
1.1 仪器与试剂
仪器设备:DRX-400型核磁共振波谱仪,Finnigan-4510型质谱仪,LC-20AT型液相色谱仪,DF-101S型集热式恒温加热磁力搅拌器,SHZ-D(Ⅲ)型循环水式真空泵,RE-52AA型旋转蒸发仪器,DZF-6050型真空干燥箱。
主要原料:4-羟基-2,2,6,6-四甲基哌啶氮氧自由基(阻聚剂701,工业级),过氧化叔丁醇(t-BuOOH)、七水合硫酸亚铁(FeSO4·7H2O)、无水亚硫酸钠(Na2SO3)、环己烷(C6H12)、乙酸乙酯(C4H8O2)、氯化钠(NaCl)(均为分析纯)。
1.2 合成工艺
各原料的摩尔比为:阻聚剂701∶C6H12∶ FeSO4·7H2O∶t-BuOOH=0.1∶1.0∶0.02∶0.5。各原料的加入量分别为:阻聚剂701 17.2g,C6H1284g,FeSO4·7H2O 6.2g,t-BuOOH 64.4g,其 中t-BuOOH的质量浓度为70%。辅料为乙酸乙酯、亚硫酸钠、氯化钠,其中Na2SO3的质量浓度为10%,NaCl的质量浓度为10%。合成反应如图2所示。
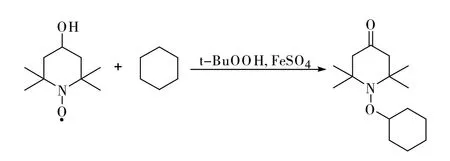
图2 1-环己氧基-2,2,6,6-四甲基哌啶-4-酮的合成反应式Fig.2 Synthesis of 1-cyclohexyloxy-2,2,6,6-tetramethylpiperid-4-one
往500mL三口烧瓶中依次加入阻聚剂701、C6H12、FeSO4·7H2O,搅拌,升温到55℃。往混合液中滴加引发剂t-BuOOH,要求在1h内滴加完毕。滴加完成后升温至65℃并在此温度下持续反应24h后,将反应体系温度降至室温。观察反应过程,可以看到溶液开始为红褐色,慢慢变为淡黄色,同时会有大量沉淀物产生,要将沉淀物过滤去除。
将200mL的C4H8O2作为稀释剂加入去掉沉淀物的滤液中,依次用Na2SO3水溶液、NaCl溶液、去离子水洗涤滤液。用Na2SO3水溶液洗涤,可将过量的t-BuOOH去除,用NaCl溶液、去离子水洗涤,可除去未反应的氮氧自由基。观察水层基本无色后,洗涤结束。在有机相中加入无水Na2SO4,再经过滤、减压蒸馏、真空干燥,即可获得1-环己氧基-2,2,6,6-四甲基哌啶-4-酮。计算得到本反应的收率为68.5%。
2 实验结果与讨论
2.1 反应机理探讨
本实验以t-BuOOH为氧化剂,FeSO4·7H2O为还原剂,构成氧化-还原引发体系,反应过程如下:Fe2+在催化剂的作用下转移至有机相,并被氧化为Fe3+,氧化剂t-BuOOH进入反应体系后,转化为自由基t-BuO·,此自由基将阻聚剂701中的羟基转化成氧,从而获得中间产物氧氮自由基。持续滴加引发剂t-BuOOH,过量的t-BuO·与C6H12发生反应,C6H12失去氢得到了自由基,上述获得的中间产物氮氧自由基与环己基自由基发生偶合反应,从而获得产品。反应机理如图3所示。
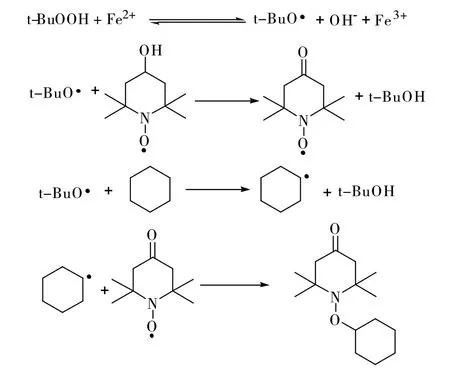
图3 1-环己氧基-4-氧-2,2,6,6-四甲基哌啶酮的反应机理Fig. 3 The mechanism of the reaction of 1-cyclohexyloxy-2,2,6,6-tetramethylpiperid-4-one
2.2 表征
将获得的产品进行了核磁共振氢谱、核磁共振碳谱、ESI-MS表征。
以CDCl3为溶剂,获得的核磁共振氢谱、核磁共振碳谱的各碳位置见图4。1H-NMR (C D C l3):3.6 9(s,1 H,C H),2.5 3(m,2 H,C H2),2.24(m,2H,CH2),2.08(s,2H,CH2),1.78(s,2H,CH2),1.4 1~1.5 8(m,6 H,C H2),1.2 8(s,6 H,C H3),1.23(s,6H,CH3)。13C-NMR(CDCl3),×10-6:208.11(1号碳),53.49(2号碳),65.42(3号碳),32.36(4号碳),85.03(5号碳),32.63(6号碳),33.77(7号碳),22.28(8号碳),20.87(9号碳),27.73(10号碳)。

图4 产品的碳谱位置图Fig.4 Carbon product position
从碳谱可以看出,由本实验获得的产品,其各碳原子的化学位移是相对应的(图5)。
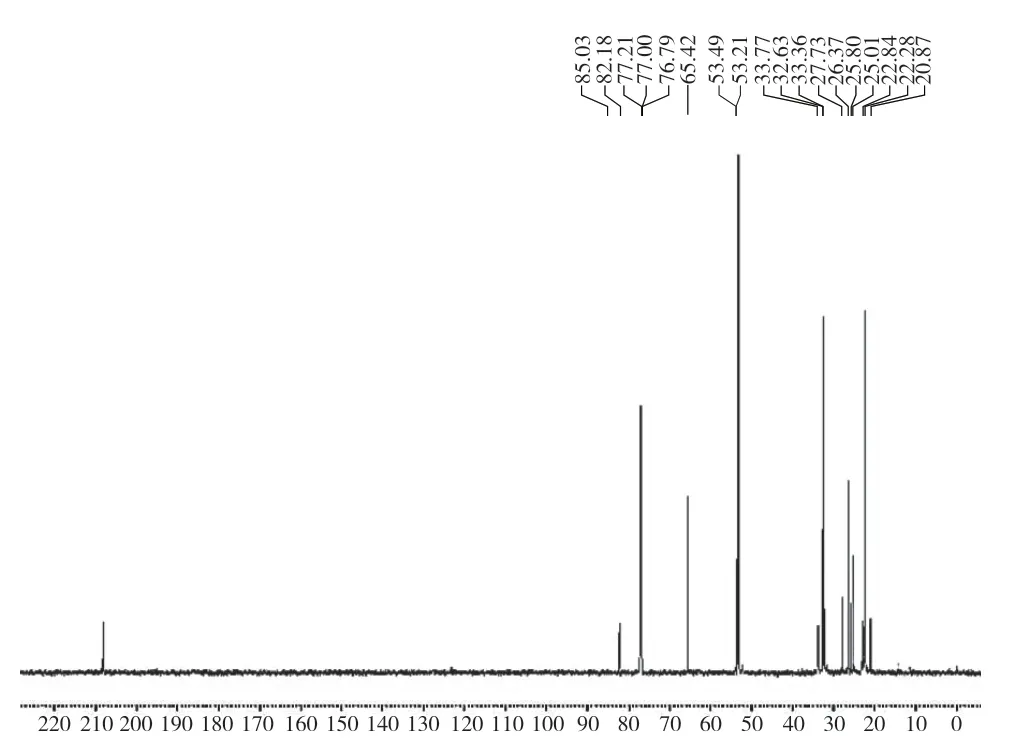
图5 1-环己氧基-4-氧-2,2,6,6四甲基哌啶的碳谱图Fig.5 13C-NMR spectra of 1-cyclohexyloxy-2,2,6,6-tetramethylpiperid-4-one
图6是1-环己氧基-4-氧-2,2,6,6四甲基哌啶的质谱图。从图6可知,ESI-MS(m/z)为254(M+,100%),与目标产品的的分子量相符。
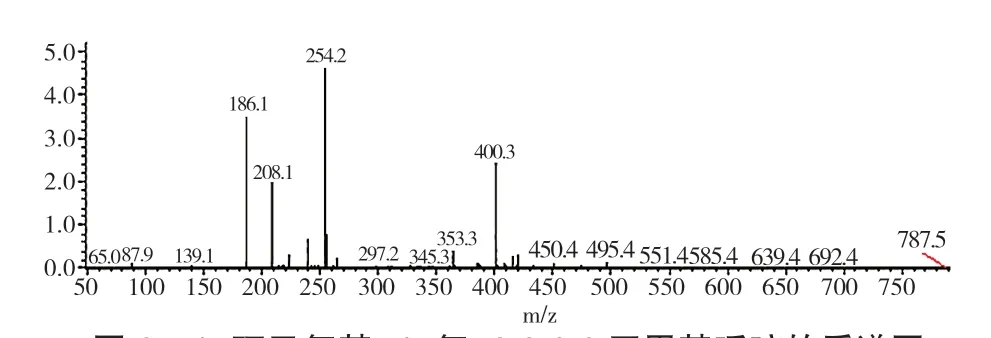
图6 1-环己氧基-4-氧-2,2,6,6四甲基哌啶的质谱图Fig.6 ESI-MS spectra of 1-cyclohexyloxy-2,2,6,6-tetramethylpiperid-4-one
从13C-NMR、质谱1、H-NMR的数据可以确定,本方法获得的产物为1-环己氧基-4-氧-2,2,6,6-四甲基哌啶酮,纯度为99.7%。
3 结论
本文以阻聚剂4-羟基-2,2,6,6-四甲基哌啶氮氧自由基(阻聚剂701)和C6H12为原料,在以过t-BuOOH为氧化剂,FeSO4·7H2O为还原剂的引发体系中,获得了自由基中间体,氮氧自由基与环己基自由基发生反应,即可获得目标产物1-环己氧基-4-氧-2,2,6,6-四甲基哌啶酮。采用核磁共振、质谱与高效液相色谱等手段,对合成产物进行了表征与纯度分析。从分析测试结果可知,采用本方法制备的中间体,收率达68.5%,纯度为99.7%,收率与纯度明显优于现有的合成技术。本方法为一步法反应,有效缩短了合成路线,反应条件简单,工艺稳定可靠,原料成本低,能够实现连续、稳定的规模化生产。
猜你喜欢
浙江化工(2024年2期)2024-03-15 02:27:40
健康体检与管理(2022年2期)2022-04-15 22:33:17
化工设计(2020年5期)2020-11-05 09:41:02
山东化工(2020年8期)2020-06-12 05:19:14
铜仁学院学报(2018年6期)2018-07-05 09:47:36
石油炼制与化工(2017年5期)2017-04-06 19:47:30
中国洗涤用品工业(2016年2期)2016-02-28 19:03:18
合成化学(2015年1期)2016-01-17 08:53:55
中国塑料(2015年2期)2015-10-14 05:34:31
云南中医学院学报(2015年2期)2015-07-31 18:11:59
