
侵染早期小麦叶锈菌相关基因的筛选及表达分析
2022-09-22 11:06张艺博路亚南肖继斌邢国珍马振玲郑文明
中国农业大学学报 2022年10期
张艺博 路亚南 刘 娜 肖继斌 李 闯 程 琨 邢国珍 马振玲 郑文明
(河南农业大学 生命科学学院/小麦玉米作物学国家重点实验室,郑州 450002)
小麦叶锈病是由叶锈菌(PucciniatriticinaEriks,Pt)引起的一种真菌气传病害,其流行性强,在世界范围内的小麦种植区均有发生,可以造成小麦严重的产量损失[1-3]。例如,2015和2016年黄淮海地区叶锈病大流行,对河南和安徽等省的小麦产区造成严重威胁[4-5]。防治叶锈病最经济有效的方法是培育小麦抗性品种,而叶锈菌毒性的变异是导致品种抗病性快速丧失的根本原因,因此揭示叶锈菌毒性/致病性相关基因对阐明叶锈菌致病机制及创制广谱抗叶锈小麦新种质具有重要意义。
叶锈菌夏孢子萌发是其侵入寄主的首要条件,了解夏孢子的萌发及早期侵染过程对小麦叶锈菌致病性的研究是至关重要的。在条件适宜的情况下,叶锈菌接触寄主2 h内便开始萌发,芽管向气孔方向延伸,接种后8 h,气孔上方可观察到大量附着胞[6],部分附着胞产生侵染栓侵入气孔,并在气孔下腔形成气孔下囊,标志着入侵成功。12 h气孔下囊分化成较短的初级入侵菌丝,接种后24 h,吸器与吸器母细胞开始形成,自此,叶锈菌夏孢子与寄主建立了寄生关系[7-8]。目前关于叶锈菌侵染早期,尚没有明确的时间定义,鉴于叶锈菌的早期侵染过程,一般认为叶锈菌侵染早期可以界定在夏孢子接触寄主24 h内。
目前,转录组测序技术已经被广泛应用于植物、动物及病原微生物等研究领域,该技术能够反映转录本的类型及数量,并在分子水平上揭示生理生化过程,从而有助于关键候选基因的挖掘。基于Illumina平台的二代转录组测序技术已应用于小麦叶锈菌分泌蛋白的预测以及差异表达基因(differentially expressed genes, DEGs)的筛选[9-10]。张悦等[11-12]利用二代转录组测序技术共预测出635个叶锈菌候选分泌蛋白,并揭示了候选基因在与寄主互作过程中的表达模式;张瑞丰等[13]通过二代转录组测序解析了叶锈菌孢子萌发的相关通路,为小麦叶锈菌致病机制的研究提供了重要基础。
随着高通量测序技术的发展,PacBio单分子实时测序技术克服了二代转录组测序技术Unigene拼接较短、转录本结构不完整的缺陷,可直接获得样本的全长转录本信息,并且在识别可变的isoform上具有更高的准确性[14]。基于PacBio平台的转录组测序已被应用于多个物种新的基因位点、isoform以及非编码RNA的鉴定[15]。已有研究者利用Illumina RNA-Seq和PacBio Iso-Seq测序技术,对植物病原真菌,如大豆锈菌等进行转录组测序[16],研究结果为揭示病原菌致病过程中的分子机制提供了依据。目前,有关小麦叶锈菌三代转录组测序分析的研究尚未见报道,本研究通过对萌发过程的叶锈菌夏孢子进行三代结合二代高通量转录组测序和表达分析,旨在筛选与叶锈菌萌发及早期侵染相关的候选基因,以期为解析小麦叶锈菌的致病分子机制提供参考。
1 材料与方法
1.1 试验材料
供试小麦品种:‘中国春’(‘CS’)、‘云麦34’(‘YM’);供试叶锈菌系:Pt-HnZU18-3单孢系由本实验室分离纯化保藏[17];3个不同毒力的叶锈菌系:PT-1,PT-2,PT-3由张艺博[18]分离鉴定所得,毒力由高到低为:PT-1>PT-2>PT-3[18]。
1.2 样品制备及RNA提取
取0.2 g叶锈菌夏孢子粉装入离心管中,加入DEPC处理的水使其充分萌发,分别制备萌发0、2、4、8、12和24 h的叶锈菌样品,采用Trizol法[19]提取RNA,样品送至北京百迈客公司进行质量检验。
1.3 测序及测序数据的处理
质量合格的样品由北京百迈客公司完成文库的构建,在PacBio RS II平台进行叶锈菌的三代转录组测序,同时构建二代文库用Illumina HiSeq平台进行转录组测序。
对PacBio RS II平台下机数据Polymerase Read进行过滤,共获得11.33 Gb的clean data,根据条件full passes≥0且序列准确性>0.75从clean data中提取ROI(reads of insert)序列,对其中的全长序列进行ICE(Iterative isoform-clustering)聚类及quiver程序纠正,利用proovread[20]软件用Illumina RNA-seq数据对低质量全长转录本进行校正。使用CD-HIT[21]软件去除冗余转录本,并用BUSCO[22]对其进行完整性评估。对Illumina HiSeq平台的下机数据进行过滤,以三代全长转录本为参考转录本,对各时间点的基因表达进行定量。
1.4 全长转录本的功能注释
使用BLAST软件将得到的非冗余转录本序列分别在NR(RefSeq non-redundant proteins)、Swiss-Prot(Swiss-Prot Protein Sequence Database)、GO(Gene Ontology)、COG(Clusters of Orthologous Groups)、KOG(euKaryotic Ortholog Groups)、Pfam(Protein family)、KEGG(Kyoto Encyclopedia of Genes and Genomes)和eggNOG(evolutionary genealogy of genes:Non-supervised Orthologous Groups)这8个数据库中进行比对,从而获得转录本的注释信息。
1.5 候选差异表达基因的筛选及生物信息学分析
通过二代转录组数据对三代转录本基因在各时间点的表达量进行差异分析,将差异倍数(Fold Change)≥2且错误发现率(False Discovery Rate,FDR)<0.01作为筛选标准。综合二代转录组差异表达分析及三代全长转录本的生物信息分析,筛选夏孢子萌发过程的重要基因。利用SignalP 4.0,TMHMM 2.0和TargetP 2.0软件对转录组中的分泌蛋白进行预测。使用NCBI保守结构域数据库NCBI保守结构域数据库(https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi)对候选基因进行结构域分析。
1.6 候选基因表达模式分析
将前期鉴定的3个不同毒力的菌系‘PT-1’、‘PT-2’、‘PT-3’分别与‘CS’(感病品种)、‘YM’(抗病品种)组成亲和与非亲和组合,取接种后0、2、4、8、12和24 h的小麦叶片,提取RNA(方法同1.2);根据候选基因的全长序列,利用Oligo 7软件进行特异性引物设计(表1),以ACTIN为内参,利用Applied Biosystems Step One Plus实时荧光定量PCR仪对候选基因进行表达分析,反应体系为:95 ℃ 10 min;95 ℃ 10 s,60 ℃ 30 s(40个循环);4 ℃保存,候选基因的相对表达水平按照2-ΔΔCT方法计算。

表1 荧光定量PCR引物(5’→3’)Table 1 Primers of qRT-PCR (5’→3’)
2 结果与分析
2.1 转录组数据统计
由表2可知,从clean data中提取441 355条ROI序列,其中包含255 706(57.9%)条全长序列、179 164(40.6%)条非全长序列和被过滤掉的6 485条<300 bp的序列,253 744条序列属于全长非嵌合序列。Illumina HiSeq平台共产出Raw data 60.191 Gb,过滤后的Clean data为58.778 Gb。由表3可知,Q20(碱基识别精度99%)达到94.98%以上,Q30(碱基识别精度99.9%)达到92.22%以上,GC含量集中在54.95%~56.50%。

表2 F01样品的ROI数据统计Table 2 ROI data statistics of F01 sample

表3 Illumina HiSeq平台产出数据统计Table 3 Summary for Illumina Hiseq sequencing
2.2 全长转录本的功能注释
由图1可知,转录本在细胞组分中主要注释到:细胞(Cell)、细胞组件(Cell Part)、细胞器(Organelle)等;在分子功能中主要注释到:催化活性(Catalytic Activity)及结合(Binding);在生物过程中主要注释到:代谢过程(Metabolic Process)与细胞过程(Cellular Process)。

图1 叶锈菌夏孢子转录本的GO注释Fig.1 GO annotation of urediniospores transcript of leaf rust
2.3 候选基因的筛选及生物信息学分析
由表4可知,相较于休眠夏孢子,萌发2、4、8、12和24 h的夏孢子分别有5 121、2 329、2 424、3 199 和3 699个差异表达基因。利用SignalP 4.0,TMHMM 2.0和TargetP 2.0软件预测出1 169个分泌蛋白;相较于休眠夏孢子,萌发2、4、8、12和24 h分别有294、123、116、205及183个差异分泌蛋白。叶锈菌的早期侵染过程中,接触寄主8和12 h分别为附着胞大量产生及形成初级菌丝的关键时期,是叶锈菌最终成功侵染并建立寄生关系的前提基础,因此,以萌发8与12 h为主要时间点,结合全长转录本的基因表达差异倍数及注释信息,筛选可能与病原菌致病力相关的基因及分泌蛋白。
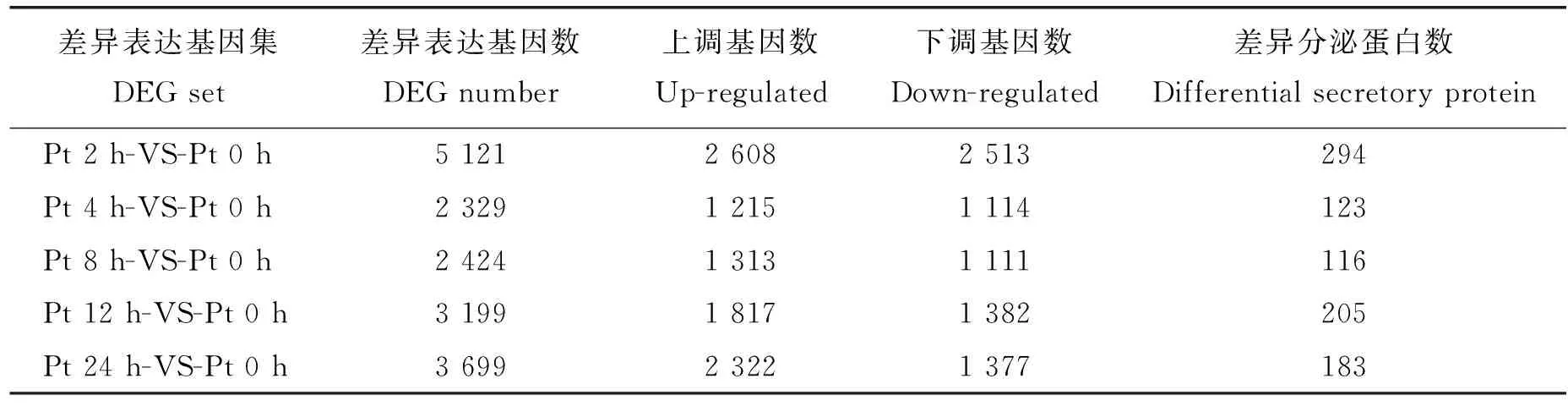
表4 叶锈菌夏孢子萌发过程差异表达基因筛选Table 4 Screening of differentially expressed genes during urediniospores germination of Puccinia triticina
由表5可知,差异表达基因的GO注释结果涉及代谢过程、ATP结合、氨基酸的生物合成、MAPK(mitogen-activated protein kinase)信号通路及氧化磷酸化等,其中还包括部分差异分泌蛋白(PTTG_1279、PTTG_3315、PTTG_1645、PTTG_1387、PTTG_2743、PTTG_1645)。结合候选基因在转录组中的表达模式及生物信息学分析,共筛选到4个与叶锈菌萌发过程相关的重要基因,分别为:PTTG_1050、PTTG_4765、PTTG_1823、PTTG_1279。

表5 部分差异表达基因列表Table 5 List of partial differentially expressed genes
由图2可知,候选基因PTTG_1050、PTTG_4765和PTTG_1279在整个萌发过程中呈现上调表达趋势,PTTG_1823为萌发过程中的下调表达基因。PTTG_1050编码292个氨基酸,全长转录组功能注释结果显示其参与SNARE(Soluble N-ethylmaleimide-sensitive factor attachment protein receptors)介导的囊泡运输,结构域注释为Syntaxin-6_N超家族、SNARE超家族。候选基因PTTG_4765编码959个氨基酸,属于ABC转运蛋白基因家族PDR亚族,结构域注释为3a01205超家族,为多向耐药性家族蛋白,见图3。PTTG_1823编码430个氨基酸,参与MAPK信号通路,属于PKc_Like(protein kinase C-like)超基因家族。候选基因PTTG_1279编码213个氨基酸,为本研究预测的分泌蛋白编码基因,其编码蛋白符合一般分泌蛋白特征:氨基酸<300个,具有信号肽,无跨膜结构域,富含半胱氨酸,未注释到保守结构域(图3)。
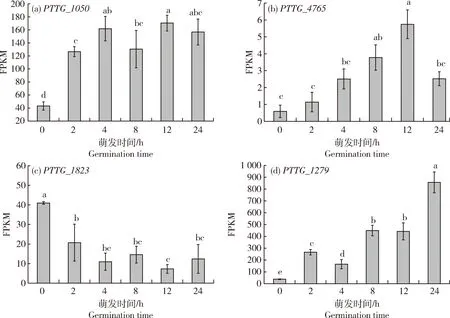
FPKM,每百万测序碱基中每千个转录子测序碱基中所包含的测序片段数。不同小写字母表示差异显著(P<0.05)。下同。FPKM represent number of sequenced fragments contained in every thousand transcripts of sequenced bases per million of sequenced bases. Different lowcase letter indicate difference significant at 0.05 level. The same below.

图3 候选基因PTTG_1050(a)、PTTG_4765(b)和PTTG_1823(c)的保守结构域注释Fig.3 Annotation of conserved domains of candidate genes PTTG_1050 (a), PTTG_4765 (b) and PTTG_1823 (c)
2.4 候选基因在亲和与非亲和组合中的表达模式
由图4和图5可知,PTTG_1050和PTTG_4765在不同毒力菌系中具有相同的表达模式,但在亲和与非亲和组合中,表达模式不同。2个候选基因均在强毒菌系‘PT-1’的亲和组合中显著上调表达;在非亲和组合中,侵染后2和12 h也出现上调表达趋势,但非亲和组合中,表达量峰值出现在侵染后12 h,而亲和组合中的表达量峰值出现在侵染后24 h,且亲和组合中候选基因的整体表达量显著高于非亲和组合,PTTG_1050与‘CS’的亲和组合中,表达量与叶锈菌毒性成显著正相关,推测PTTG_1050可能与叶锈菌的强毒性相关,并在叶锈菌生长发育中发挥重要作用。PTTG_4765在亲和组合中有着较高的表达量,且与非亲和组合出现表达量峰值的时间有所差异,其在亲和组合中侵染24 h达到表达量峰值,且表达量显著高于非亲和组合,推测PTTG_4765可能与叶锈菌致病力相关。
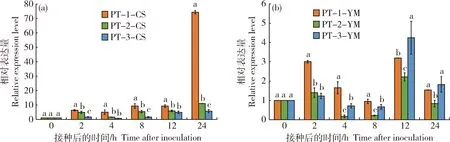
PT-1-CS、PT-2-CS、PT-3-CS分别表示‘PT-1’、‘PT-2’、‘PT-3’接种CS;PT-1-YM、PT-2-YM、PT-3-YM分别表示‘PT-1’、‘PT-2’、‘PT-3’接种YM,下同。PT-1-CS, PT-2-CS and PT-3-CS represent ‘PT-1’, ‘PT-2’ and ‘PT-3’ are inoculated with CS,respectively. PT-1-YM, PT-2-YM and PT-3-YM represent ‘PT-1’, ‘PT-2’ and ‘PT-3’ are inoculated with YM, respectively. The same below.

图5 3个不同毒力菌系与‘CS’/‘YM’的亲和(a)、非亲和(b)组合中PTTG_4765的表达Fig.5 Expression of PTTG_4765 in ‘CS’/‘YM’ compatible (a) and incompatible (b) combinations of three different virulent isolates
由图6和图7可知,PTTG_1823和PTTG_1279分别在不同毒力菌系的早期侵染过程中表现出显著不同的表达模式。PTTG_1823在亲和组合中整体为上调表达趋势,而在‘PT-1’、‘PT-2’的非亲和组合中下调表达,在‘PT-3’的非亲和组合中,侵染后2和12 h出现表达量上调,但上调倍数不超过1.6倍,表明其在抗病寄主中表达受到抑制,显示其可能与感病寄主中菌丝的顺利扩展有密切关系,与叶锈菌的毒性相关。PTTG_1279在菌系‘PT-1’与‘PT-3’中有着相似的表达模式,即在亲和组合中,侵染后2和24 h均出现表达量上调,不同的是,侵染4~8 h,其在‘PT-1’中,表达量持续下调,而在‘PT-3’中持续上调,非亲和组合中,PTTG_1279在‘PT-1’和‘PT-3’这两个菌系中的表达模式完全一致,侵染4 h出现表达量峰值;其在中度毒力菌系‘PT-2’中,有着显著不同于‘PT-1’和‘PT-3’的表达模式,即在亲和与非亲和组合中均显著上调表达,在亲和组合中,侵染8 h达到表达量峰值,后续时间点持续高表达,而在‘PT-2’的非亲和组合中,12 h达到峰值后又迅速下降,表明其在中度毒力菌系‘PT-2’中可能存在不同的作用途径或机制。

图6 3个不同毒力菌系与‘CS’/‘YM’的亲和(a)、非亲和(b)组合中PTTG_1823的表达Fig.6 Expression of PTTG_1823 in ‘CS’/‘YM’ compatible (a) and incompatible (b) combinations of three different virulent isolates
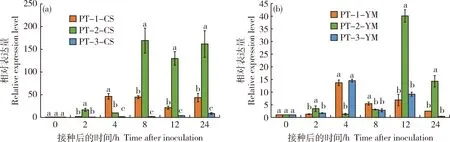
图7 3个不同毒力菌系与‘CS’/‘YM’的亲和(a)、非亲和(b)组合中PTTG_1279的表达Fig.7 Expression of PTTG_1279 in ‘CS’/‘YM’ compatible (a) and incompatible (b) combinations of three different virulent isolates
3 讨 论
作物病原真菌与寄主间的互作研究已有报道[23-24]。病原菌通过病原相关分子模式(pathogen-associated molecule pattern,PAMP)或吸器向植物体内释放效应蛋白抑制寄主免疫系统,入侵寄主植物。寄主的PAMP触发免疫反应(PAMP-triggered immunity,PTI)和效应蛋白触发免疫反应(Effector-triggered immunity,ETI)两大免疫系统相辅相成,为植物抵御病原菌的入侵提供有力保障[25-26],而叶锈菌夏孢子萌发是其侵染早期的重要发育阶段,萌发过程相关的一些关键基因在叶锈菌与小麦的早期互作及成功侵染中发挥着至关重要的作用。
本研究筛选到了叶锈菌萌发过程相关的4个关键基因,其中,PTTG_1050可能参与SNARE介导的囊泡运输,该通路被报道可能与孢子萌发相关[13],SNARE蛋白家族成员Sec22先后在稻瘟病菌[27]、大丽轮枝菌[28]以及禾谷镰刀菌[29]中被证实与病原菌致病力相关;PTTG_4765属于ABC转运蛋白基因家族PDR亚族,ABC转运蛋白能在增强病原菌抵抗不利环境中起作用[30],禾谷镰刀菌中的ABC转运蛋白基因FgATM1被报道在病原菌的致病力方面发挥关键作用[31];PTTG_1823参与MAPK信号通路,真菌的MAPK通路在菌丝侵染、附着胞形成以及致病毒力方面均起到非常重要的作用,能促进病原真菌的侵染、帮助病原真菌突破植物防御反应[32-33],同时,PTTG_1823与叶锈菌、条锈菌的致病基因PtMAPK1和PsMAPK1同属于PKc_Like超家族[34-35];PTTG_1279为本研究预测的分泌蛋白编码基因,病原菌分泌蛋白被报道能够抑制宿主的免疫反应,介导病原菌的营养吸收,进而促进病原菌的寄生与定植[36-37]。小麦叶锈菌萌发相关基因的研究可为揭示其致病分子机制,特别是为PTI相关基因的作用机制提供重要基础。
4 结 论
对萌发的小麦叶锈菌夏孢子进行了全长转录组测序,获得20 853个高质量转录本,平均长度为2 170.51 bp。将转录本在8个数据库中进行比对,97%的转录本均得到注释,其中,12 202条转录本被注释到GO数据库中。在叶锈菌夏孢子萌发2、4、8、12和24 h分别鉴定出5 121、2 329、2 424、3 199、3 699个差异表达基因,共预测出1 169个分泌蛋白,差异表达分析显示,相较于休眠夏孢子,萌发2、4、8、12和24 h分别有294、123、116、205及183个差异表达的分泌蛋白。
以萌发8与12 h为主要时间点,筛选到了4个与侵染早期叶锈菌萌发相关的重要基因,PTTG_1050为SNARE蛋白的编码基因;PTTG_4765属于ABC转运蛋白基因家族PDR亚族;PTTG_1823属于PKc_Like超基因家族;PTTG_1279为预测的候选分泌蛋白编码基因。4个候选基因在亲和互作中表达量均高于非亲和互作,表明候选基因在叶锈菌的早期侵染和扩展中发挥着重要作用。
猜你喜欢
中国蔬菜(2022年7期)2022-07-21
天津农业科学(2022年5期)2022-05-31
江苏农业科学(2019年14期)2019-09-23
中国瓜菜(2019年8期)2019-09-19
江苏农业科学(2017年19期)2017-11-22
湖北农业科学(2017年4期)2017-03-28
江苏农业科学(2016年11期)2017-03-21
江苏农业科学(2017年1期)2017-02-27
科学大众(中学)(2015年9期)2015-10-12
少儿科学周刊·少年版(2015年3期)2015-07-07
