
ADH1B基因甲基化对卵巢癌细胞增殖及凋亡的影响
2022-09-21 06:34季维雪李泽莲
安徽医科大学学报 2022年8期
季维雪,孙 磊, 江 玉, 陈 颍, 李泽莲, 李 敏, 肖 兰
作者单位:1安徽医科大学附属阜阳医院妇产科,阜阳 2361122安徽医科大学第一附属医院妇产科,合肥 230022
卵巢癌是女性生殖系统常见三大恶性肿瘤之一,病死率居首位,化疗耐药是导致卵巢癌疗效不佳的重要因素之一[1]。越来越多研究[2]显示卵巢癌发生、发展及耐药与表观遗传学改变密切相关。表观遗传修饰包括DNA甲基化、组蛋白修饰等,其中DNA甲基化异常在卵巢癌耐药形成中起着重要作用。文献[3]报道,通过DNA甲基化抑制剂逆转 DNA甲基化状态,为靶向治疗卵巢癌提供了一个新的靶点。酒精脱氢酶(alcohol dehydrogenase, ADH)是一种多态性酶,乙醇脱氢酶1B (alcohol dehydrogenase 1B, ADH1B)也称为ADH2,目前ADH1B与癌症的联系主要集中在酒精代谢和饮酒行为等方面[4]。新近研究[5]表明甲基化下调的ADH2与乳腺癌不良预后相关,人肿瘤细胞中ADH2基因亦受表观遗传机制调控,提示ADH2基因有望成为潜在肿瘤个体化治疗靶点。该研究拟体外观察5-Aza-dc对卵巢癌细胞增殖和凋亡的影响,旨在探讨ADH1B基因甲基化与卵巢癌发生发展的关系。
1 材料与方法
1.1 材料与试剂细胞株:卵巢癌细胞C13K及OV2008为本室保存;组织样本选取2019年1月—2021年5月就诊于医院妇产科且均经病理学诊断卵巢癌患者55例,FIGO分期:Ⅰ~Ⅱ期18例,Ⅲ~Ⅳ期37例。同期因子宫肌瘤需手术患者21例(留取正常卵巢上皮)。卵巢癌组患者的平均年龄(50.4±9.6)岁,对照组平均年龄(44.6±7.3)岁。排除自身免疫病及其他系统肿瘤,术前均未接受新辅助化疗、免疫及靶向治疗,经手术切除的新鲜组织标本均在30 min内。研究对象均签署知情同意书,并通过医院伦理委员会审查。DNA纯化试剂盒购自德国Qiagen公司;去甲基化制剂5-Aza-dc购自美国Sigma公司;PRMI Medium 1640培养基、小牛血清均购自美国Gibco公司;ADH1B兔多克隆抗体购自英国Abcam公司;Hoechst 33258染色液购自上海碧云天公司;Trizol试剂购自美国Invitrogen公司;逆转录试剂盒及SYBR Green Master Mix购自日本TaKaRa公司;CCK-8检测试剂盒购自上海碧云天公司。
1.2 方法
1.2.1生物信息学分析 生物信息学分析网站GEPIA(http://gepia.cancer-pku.cn/)分析ADH1B在卵巢癌的表达,数据来源于癌症和肿瘤基因图谱(cancer genome atlas,TCGA)。
1.2.2细胞培养及干预分组 C13K和OV2008细胞于含15%小牛血清1640培养液,置37℃、5% CO2培养箱培养。当细胞融合度达(70~80)%时,0.25%胰蛋白酶消化传代,取对数生长期细胞进行实验。5-Aza-dc设低、中及高3个浓度组,分别为0.5、2.5及10 μmol/L,分别干预48 h。
1.2.3去甲基化药物对卵巢癌细胞毒性作用 各组细胞按1×104细胞/孔浓度种至96孔板,继续培养24 h。细胞增殖:5-Aza-dc (0.5、2.5及10 μmol/L)干预48 h,加10 μl CCK-8溶液,继续培养4 h,450 nm波长处各孔吸光度值。实验均重复3次,取平均值。
1.2.4Hoechst33258染色法观察细胞形态变化 两株细胞以2×105细胞/孔浓度接种于6孔培养板,细胞贴壁后加高浓度5-Aza-dc(10 μmol/L)继续培养48 h,PBS洗涤细胞2次,4%多聚甲醛固定30 min,PBS洗涤细胞2次,Hoechst 33258(5 mg /L)在室温下染色10 min后弃去染色液,PBS洗2遍,荧光显微镜下观察,拍照。正常细胞核Hoechst着色形态呈圆形,淡蓝色;凋亡细胞核浓集固缩而呈亮蓝色。
1.2.5甲基化特异性PCR (methylation specific PCR, MSP) 采用DNA甲基化修饰试剂盒对基因组DNA进行甲基化修饰,具体操作按试剂盒说明书进行。PCR反应条件:95 ℃预变性12 min;94 ℃变性30 s,退火温度58 ℃,72 ℃延伸45 s,45个循环;72 ℃延伸10 min。扩增后产物用2%琼脂糖凝胶电泳,凝胶成像系统观察分析。ADH1B MSP甲基化引物序列F:5′-CCAGGGATTAGGAGTGGACC-3′,R: 5′-GGAGGGGAAGAGCAGTTGTC-3′; 未甲基化引物序列, UF: 5′-CAGTGTGGAAAATGCAGAG-3′; UR:5′-GTGACCTTGGCAACGTTA-3′。MSP结果判定:仅扩增出甲基化条带者为完全甲基化;同时出现甲基化条带和非甲基化条带者为部分甲基化; 仅出现非甲基化条带者为未甲基化。
1.2.6qRT-PCR检测ADH1BmRNA水平 收集组织标本及5-Aza-dc(0.5、10 μmol/L)作用两组细胞,将组织样本置液氮中研磨成匀浆,TRIzol法提取组织和细胞RNA,反转录合成cDNA。PCR引物序列,ADH1B,F: 5′-GTGGCACAAGCGTCATCGTAGG-3′,R: 5′-TTCCAGGTGCGTCCAGTCAGTAG-3′;β-actin,F: 5′-AGAAGGCTGGGGCTCATTTG-3′,R: 5′-AGGGGCCATCCACAGTCTTC-3′。2-△△Ct法计算各组细胞中ADH1B基因mRNA相对表达量,实验重复3次。
1.2.7免疫印迹检测ADH1B蛋白水平 30 μg蛋白质样品行SDS-PAGE电泳,湿转至硝酸纤维膜上;5% BSA室温封闭2 h,0.05% Tween20的TBS缓冲液(TBST)漂洗,10 min×3次;加入ADH1B(1 ∶1 000)一抗,b-actin一抗(1 ∶5 000),4 ℃过夜,1×TBST漂洗3遍,对应辣根过氧化物酶标记二抗(1 ∶5 000),37 ℃摇床温育2 h,ECL显色曝光,采集照片。

2 结果
2.1 ADH1B在卵巢癌中表达被抑制为明确ADH1B在OC中表达情况,根据TCGA数据中ADH1B在426例OC患者样本和88例正常样本表达的差异结果显示,ADH1B在肿瘤样本中比正常样本低,差异有统计学意义(P<0.05) , 见图1。
2.2 组织及两株细胞中ADH1B基因表达qRT-PCR证实,正常卵巢上皮组织中ADH1B表达水平为(6.72±1.41),高于卵巢癌组织(1.15±0.67),差异有统计学意义(t=-5.742,P<0.01);OV2008细胞ADH1B基因表达为C13K细胞5.82倍,差异有统计学意义(t=-7.313,P<0.01)。
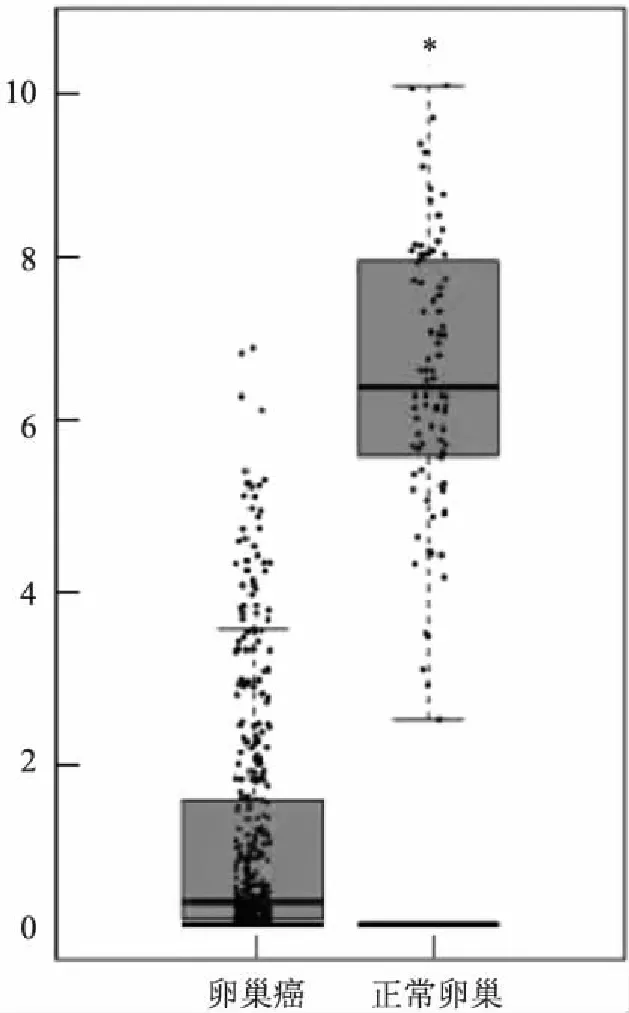
图1 ADH1B 在卵巢癌及正常卵巢上皮组织中相对表达量
2.3 5-Aza-dc对细胞存活率比较低浓度5-Aza-dc(0.5 μmol/L)对两株卵巢癌细胞生长无明显抑制作用(P>0.05);中浓度5-Aza-dc(2.5 μmol/L)对两株卵巢癌细胞生长有一定抑制作用(t=9.234,P<0.01;t=15.416,P<0.01),当5-Aza-dc增至高浓度10μmol/L时,两株细胞生长抑制差异显著(F=40.082,P<0.001;F=80.384,P<0.001)(表1)。
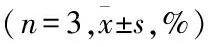
表1 各组细胞存活率比较
2.4 细胞凋亡形态学的变化荧光显微镜下,经高浓度5-Aza-dc(10 μmol/L)作用48 h后,OV2008及C13K细胞均出现典型细胞凋亡形态学改变,表现为核染色质浓缩、碎裂,而未加药两株对照细胞核均呈均匀蓝色荧光,见图2。

图2 两株细胞细胞凋亡Hoechst 33258荧光染色 ×200
2.5 5-Aza-dC对ADH1B启动子甲基化影响结果显示:ADH1B基因启动子区在OV2008及SKOV3细胞株中呈完全甲基化;高浓度5-Aza-dC (10 μmol/L)处理48 h后,可见ADH1B基因启动子区出现U带,M带未完全消失,提示ADH1B启动子区甲基化被部分逆转,见图3。

图3 5-Aza-dC处理前后两株细胞ADH1B基因启动子甲基化
2.6 5-Aza-dC对ADH1B mRNA表达影响OV2008细胞中ADH1B mRNA表达高于C13K细胞;5-Aza-dC(0.5、10 μmol/L)作用48 h后,OV2008及C13K细胞ADH1B mRNA均有所升高,尤以高浓度5-Aza-dC(10 μmol/L)处理后OV2008及C13K细胞ADH1B mRNA升高差异显著(F=8.093,P<0.01;F=21.928,P<0.01),图4。
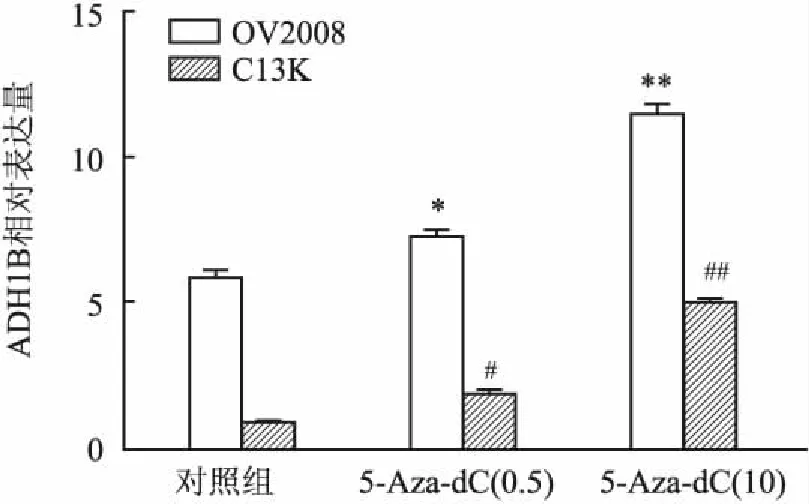
图4 细胞中ADH1B mRNA表达水平比较
2.7 免疫印迹检测ADH1B蛋白水平结果显示,无5-Aza-dC作用,OV2008及C13K细胞中ADH1B蛋白呈较低表达状态,且C13K中表达较之OV2008更低;低浓度5-Aza-dC(0.5 μmol/L)未引起OV2008及C13K细胞中ADH1B蛋白改变,而高浓度5-Aza-dC(10 μmol/L)使OV2008及C13K细胞中ADH1B蛋白均表达增加,见图5A。ADH1B/b-actin对蛋白图像扫描半定量统计结果表明,低浓度5-Aza-dC(0.5μmol/L)处理后,两株细胞ADH1B蛋白表达无明显改变(P>0.05);高浓度5-Aza-dC (10 μmol/L)处理使ADH1B蛋白表达较两株对照组及低浓度组升高,差异有显著性(F=12.059,P<0.01;F=30.384,P<0.01),见图5B。
图5 各组细胞ADH1B蛋白表达
3 讨论
ADH1B基因编码 I 类ADH的β亚基,定位于染色体4q23。Gharpure et al[6]发现ADH1B异常表达使卵巢癌细胞分泌金属基质蛋白酶7、白细胞分化抗原26和组织蛋白酶从而促进肿瘤进展。ADH1B在鼻咽癌细胞中表达下调,鼻咽癌细胞过表达ADH1B后可抑制鼻咽癌细胞的增殖和迁移能力,逆转细胞的恶性程度[7]。另有研究[8]指出ADH1B可能为卵巢癌细胞化疗耐药的候选基因之一。本研究qRT-PCR结果及卵巢癌TCGA数据分析均提示ADH1B在OC患者样本中低表达,据此课题组推测ADH1B基因可能作为抑癌基因参与了卵巢癌的发生发展,但其具体调控机制目前尚未明确,有必要进行深入探讨。
肿瘤细胞DNA异常甲基化是一种常见表观遗传学改变,这种异常修饰可抑制基因转录,使抑癌基因丧失功能[9-10]。基因启动子区DNA甲基化是导致基因失活的重要原因之一,而基因启动子甲基化的潜在可逆性为新型抗癌药物的开发提供了机会,这些药物可重新激活沉默的肿瘤抑制基因[11]。以5-Aza-dc为代表的类核苷甲基转移酶抑制剂,通过与DNA甲基转移酶共价结合抑制其活性,降低基因甲基化水平,已广泛应用于逆转肿瘤细胞的异常甲基化,诱导因甲基化而沉默的抑癌基因重新表达,抑制肿瘤细胞生长,从而达到杀伤肿瘤细胞的效应[12-13]。为探讨ADH1B基因甲基化与卵巢癌发生发展的关系,本研究将 5-Aza-dC作用于两株卵巢癌细胞株,通过CCK-8 法检测5-Aza-dc对人卵巢癌细胞生长的影响,结果表明,5-Aza-dc对两株卵巢癌细胞增殖均有抑制作用,且该作用与 5-Aza-dc浓度呈正相关;同时,Hoechst 33258荧光染色结果显示,高浓度5-Aza-dc处理后,细胞核染色质浓缩或碎裂,出现典型细胞凋亡形态,表明5-Aza-dc诱导卵巢癌细胞发生凋亡;RT-PCR结果显示,ADH1B mRNA表达与启动子区甲基化程度密切相关,在完全甲基化两株卵巢癌细胞中,ADH1B mRNA表达较低,而5-Aza-dC作用48 h后,两株细胞ADH1B甲基化均被部分逆转,ADH1B在mRNA和蛋白水平表达均显著提高,该结果提示5-Aza-dC可能通过沉默DNA甲基转移酶而降低抑癌基因ADH1B 启动子区域的甲基化水平诱导其重新表达。
综上所述,ADH1B基因甲基化在OC发生发展中发挥重要作用,5-Aza-dc可部分逆转卵巢癌细胞启动子区ADH1B甲基化,诱导ADH1B基因重新表达,抑制肿瘤生长,促进细胞凋亡,从而发挥抗肿瘤效应。本研究也为将5-Aza-dc应用于卵巢癌临床治疗提供一定的理论依据。
猜你喜欢
上海师范大学学报·自然科学版(2022年3期)2022-07-11
中国农学通报(2022年13期)2022-05-31
作物学报(2022年5期)2022-03-16
伴侣(2021年6期)2021-08-03
家庭百事通·健康一点通(2020年12期)2020-12-31
健康之友(2020年1期)2020-03-24
绿色科技(2017年8期)2017-05-22
绿色科技(2017年2期)2017-03-23
中国民族民间医药·下半月(2014年4期)2014-09-26
祝你幸福·知心(2008年1期)2008-12-29
