
微筛孔反应器液相沉淀法制备纳米CaCO3
2022-09-14 11:32朱子玉王豪波叶文财曹建新
人工晶体学报 2022年8期
朱子玉,宣 超,王豪波,叶文财,易 芸,曹建新
(1.贵州大学化学与化工学院,贵阳 550025;2.贵州省绿色化工与清洁能源技术重点实验室,贵阳 550025; 3.贵州省工业废弃物高效利用工程研究中心,贵阳 550025)
0 引 言
冷冻法硝酸磷肥工艺不仅不产生固废磷石膏,还使复合肥料含有更易于植物吸收的硝态氮。但冷冻法硝酸磷肥生产过程中,每吨氮磷肥(NP)约副产0.6 t Ca(NO3)2·4H2O,目前Ca(NO3)2·4H2O的加工利用比较单一,主要用于生产硝酸铵钙产品,而硝酸铵钙具有的易吸潮结块、单位养分价格高[1]、钙含量远高于农作物需求等问题影响其发展应用,因此,Ca(NO3)2·4H2O在一定程度上成为制约冷冻法硝酸磷肥技术发展的一个瓶颈,而拓展Ca(NO3)2·4H2O的加工技术和良好应用也一直是一个难题。结合冷冻法硝酸磷肥工艺特点和市场需求,可以考虑用(NH4)2CO3和Ca(NO3)2·4H2O为原料制备CaCO3[2]。
但传统方法制备CaCO3所需设备庞大、混合效率低,且需通过多种复合外加剂,以及繁琐的操作来调控晶粒、晶相与形貌[3-5],相比之下,微流控技术因具备传质速率高、瞬时混合、分散性强、装置小且无放大效应等优势[6],可有效解决传统方法的问题[7-8],已然成为纳米粉体制备领域关注的热点工艺技术。但对于液相沉淀反应体系,微流控技术存在反应器易堵塞的问题[9],为了解决堵塞问题,根据反应体系的特点可以考虑使用气相反应物料或引入惰性气体介质强化体系扰动[10],借助超声等外力作用清除堵塞物[11],设计特殊微通道结构强化液相混合过程以促进反应体系分散[12-14]等。微筛孔反应器具有独特的孔道阵列结构,可将分散相流体以微米级气泡或液滴的形式分散至连续相流体中,使两种流体在微通道内以分子扩散的形式进行混合,从而增大体系的过饱和度、均匀度与分散性[16];同时该微反应器的错流空间结构还使连续相对分散相物料产生剪切作用,有力推动反应物料的流动,有效缓解一般微反应器出现的堵塞问题。为此,本文选用微筛孔反应器,并针对(NH4)2CO3和Ca(NO3)2·4H2O液相沉淀反应特性和产物性质适当调整微筛孔孔径及排布,以分析纯Ca(NO3)2·4H2O为钙源连续相、(NH4)2CO3为碳源分散相,研究常温常压条件下微反应器技术于液相沉淀体系制备纳米碳酸钙晶体的可能性,借助 XRD、TEM等表征手段重点研究了连续相与分散相流量、物料浓度以及停留时间等工艺条件对样品晶相、产率、形貌、粒径及分布的影响。以期为实现实际工业生产提供一定的参考。
1 实 验
1.1 实验原料与制备方法
Ca(NO3)2·4H2O (纯度99.0%,AR,成都金山化学试剂有限公司);(NH4)2CO3(纯度99.0%,AR,天津市科密欧化学试剂有限公司);NH4H2PO4(纯度99.8%,GR,天津市科密欧化学试剂有限公司)。
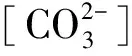
1.2 性能测试与表征
使用FEI Tecnai F20透射电子显微镜(TEM)测量样品尺寸并通过数字显微软件Image Pro基于统计数字加权法在TEM照片上测量250~300个粒子进行量化分析;采用Bruker AXS GmbH D8 Advance X射线粉末衍射(XRD)仪分析样品的晶相组成(工作条件为Cu Kα、电压40 kV、电流40 mA、扫描速度20 (°)/min、扫描范围2θ=20°~60°、X射线波长λ=0.154 06 nm);采用Scherrer方程计算晶体的平均粒径。
CaCO3产率Y以式(1)计算:
(1)
式中:M1是运行微反应器1 min后制备出的CaCO3实际质量,g。M是运行微反应器1 min制备CaCO3的理论质量,g。
2 结果与讨论
2.1 制备参数对样品晶相及产率的影响
2.1.1 连续相和分散相流量对样品晶相及产率的影响
当FC=50 mL/min,FD=50 mL/min,τ=25 s时,连续相和分散相流量对样品晶相及产率影响的实验结果如图2所示。由图2(a)、(b)可见,FD为50 mL/min时,FC从50 mL/min变化至150 mL/min所制得样品的XRD图谱与方解石的标准卡片PDF#05-0586一致,晶相均为方解石,且衍射峰强度随着FC增大而逐渐减弱(见图2(a)),而产率自82.57%(质量分数,下同)上升至95.89%(见图2(b))。这是因为在不改变温度与压力的条件下,FC不断提高时错流微通道(混合腔)内的连续相拖曳力也在不断增大,强化了微反应器内的扰动并加剧了两相物料的混合程度,同时微通道单位体积内的Ca(NO3)2量随FC增大而上升,致使体系过饱和度提升[17],而过饱和度又与成核速率呈正相关,即过饱和度越大成核速率越高,微通道内瞬时成核量越大[8],产率随之增大,而衍射峰强度逐渐减弱则是两相溶液混合扰动程度的加剧和过饱和度的提升,影响方解石晶体稳定生长所致,晶体的结晶度有所降低,在图谱中表现为半峰全宽变大。
由图2(c)、(d)可见,FC为50 mL/min时,FD从50 mL/min变化至150 mL/min,所制得样品的晶相与衍射峰强度变化趋势与FC增大时的实验结果一致,但产率先随FD从50 mL/min增大至100 mL/min的过程由82.57%上升至94.66%,后随FD增至150 mL/min的过程逐渐降至93.76%。这是因为分散相进料流量的增大,同样会使微通道内(NH4)2CO3的物料量增加,连续相与分散相物料的混合程度加剧使得体系过饱和度提升,故FD增大产率上升,衍射峰强度减弱。但FD过高时有可能影响连续相与分散相物料流的有效混合与反应,致使产率略有下降。
2.1.2 连续相和分散相浓度对样品晶相及产率的影响
当FC=50 mL/min,FD=50 mL/min,τ=25 s时,连续相与分散相浓度对样品晶相及产率影响的实验结果如图3所示。
2.1.3 停留时间对样品晶相及产率的影响
由图可见,反应生成的晶相均为方解石,而XRD峰强度总体随停留时间增加呈现逐渐增强趋势(除1 s条件下制备的样品,见图4(a)),同时与方解石标准卡片对比可发现,停留时间低于15 s时所制备样品的(006)晶面衍射峰出现了向右偏移的现象;产率则在停留时间从1 s增加至5 s时从91.82%提升到了94.27%,之后持续下降,停留时间20 s时产率降至82.57%(见图4(b))。
停留时间主要影响连续相和分散相物料的混合反应与晶体生长过程。由于停留时间是通过外接微反应管的长度进行调整的,停留时间越长微反应管也就越长,晶体在管内生长也越充分,故而衍射峰强度呈现渐强趋势,样品结晶度提升,衍射峰半峰全宽减小;同时,停留时间低于15 s时,晶体的(006)晶面生长受到影响致使衍射峰出现偏移[18]。微反应管越长晶体在管内壁的粘附损失增大,故停留时间过长产率反而减小[17]。而τ=1 s时射峰强度相对较高,可能是因为停留时间过短,两相物料混合只能形成极少晶核并得以在连续相容器内生长。
2.2 制备参数对样品粒径和形貌的影响
2.2.1 连续相和分散相流量对样品粒径和形貌的影响
图5为[Ca2+]=0.05 mol/L,[CO32-]=0.05 mol/L,τ=20时不同物相流量制备样品的平均粒径图。从图中可以看出:平均粒径随FC提升从78.79 nm逐渐降至51.58 nm(见图5(a));平均粒径随FD提升从78.79 nm不断减小至50.90 nm(见图5(b))。由此可看出连续相与分散相流量提升时,Scherrer公式计算的样品晶体平均粒径dXRD均呈现减小趋势。这是因为FC提升时,连续相的剪切力与传质面积随之增大,减小了分散相的几何尺寸,导致晶体生长受阻、粒径减小,这与文献[19-20]实验结论一致;而FD提升时,分散相进入连续相的扩散通量与传质效率增大,两相混合程度加剧,从而使得过饱和度升高,晶体粒径减小[8]。
反应条件为FC=FD=50 mL/min,FC=150 mL/min、FD=50 mL/min,FC=50 mL/min、FD=150 mL/min时制备的3个样品TEM照片及粒径分布如图6所示。
以上实验结果表明,本实验条件下较为适宜的两相流量为:FC=50 mL/min、FD=150 mL/min。
2.2.2 连续相和分散相浓度对样品形貌及粒径的影响
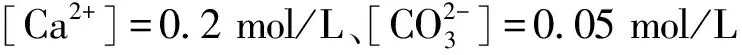

2.2.3 停留时间对样品形貌及粒径分布的影响
从图11可以看出,随停留时间延长,样品的dXRD先从67.92 nm缩小到了47.53 nm(1~5 s),之后持续上升,至20 s时达到78.79 nm。原因在于停留时间过短会使得反应物料在微通道内混合反应不完全、晶核相对较少,晶体的生长主要发生在连续相容器中,粒径较大;而停留时间过长则会使晶体在管内过度生长从而导致粒径增大[24];只有适宜的停留时间能使两相物料得以充分混合反应并结晶。
3 结 论
(1)本文采用微筛孔反应器以液相沉淀法制备了纳米碳酸钙,通过XRD、TEM表征发现不同制备条件下均能合成方解石碳酸钙,其产率保持在80%以上。同时,在具体实验中发现微筛孔反应器特有的混合模式与分散特点有效避免了堵塞现象的发生,可实现连续化生产。
(2)两相进料流量增大时,样品的粒径均有减小且分布变窄。但产率随连续相流量升高而增大;随着分散相流量的增加却呈现先增大后减小的趋势,FC=50 mL/min、FD=150 mL/min分别是连续相和分散相流量单因素实验中的最佳参数。
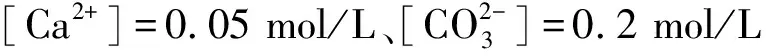
(4)除τ=1 s外,样品产率均随停留时间延长而减小,粒径增大,而停留时间过短(τ=1 s)制得的样品粒径都很大且分布较宽;τ=5 s是最为适宜的停留时间。
猜你喜欢
可再生能源(2022年5期)2022-06-09
节能与环保(2022年4期)2022-06-02
城市道桥与防洪(2022年3期)2022-05-08
中国食用菌(2021年10期)2021-11-04
湖北农机化(2021年11期)2021-07-01
安全与环境工程(2021年2期)2021-04-02
昆钢科技(2021年6期)2021-03-09
汽车零部件(2020年9期)2020-09-27
北京航空航天大学学报(2017年2期)2017-11-24
北京航空航天大学学报(2017年2期)2017-11-24
