
RNA m6A-Methylation in Neurodevelopment
2022-09-07 03:59XUELiTingZHAOTianYEHaiHong
中国生物化学与分子生物学报 2022年3期
XUE Li-Ting, ZHAO Tian, YE Hai-Hong*
(1)Department of Medical Genetics and Developmental Biology, School of Basic Medical Sciences, Capital Medical University, Beijing 100069,China;2)Beijing Key Laboratory of Neural Regeneration and Repair, Capital Medical University, Beijing 100069, China)
Abstract The central nervous system controls high-level neural activities such as perception, movement, language, and cognition. As the most important part of the human nervous system, its normal development and functional activities are very important in the process of human development. A better understanding of the essential molecular pathways that regulate the development of the nervous system may improve diagnoses and treatments for neurologic diseases, as well as basic biological understanding of the brain. The dynamic changes of the modified state of RNA N6-methyladenosine (m6A) and its functions are mainly regulated by m6A methyltransferase, m6A demethylase and m6A reading protein complexes, which are described in detail in this paper. Herein, we introduce the molecules involved in the methylation process of m6A in detail. We also provide a detailed summary of the effects of m6A modifications on neurodevelopment, with a focus on the role of epi-transcriptomics in gene regulation, including neurogenesis, neural differentiation, axon guidance, synaptic formation and synaptic plasticity. Further, we emphasize the biological significance of m6A modifications in neurons during development. We also introduce several methods for detecting the m6A site based on different experimental principles and technique. Each method has its own merits and we will be able to study the modification both more broadly and in greater depth. We also can choose the appropriate method to process our projects. In recent years, with the development of m6A sites detection technology, the role of m6A methylation in the development of the nervous system and the occurrence of neurological diseases has gradually become a hot topic. RNA m6A methylation is a new frontier in neuroscience with clear potential to provide exciting new perspectives on neurodevelopment and neurological diseases.
Key words epi-transcriptomics; N6-methyladenosine (m6A); neurodevelopment; neurogenesis; synapse
The nervous system is precisely regulated by different mechanisms to ensure proper function, and the process of neurodevelopment requires dynamic changes in gene expression to regulate cell fates. Unsurprisingly, abnormalities in neurodevelopment are associated with neurological diseases[1,2]. The central dogma of molecular biology asserts that DNA sequences are transcribed into RNAs, which are translated into functional proteins[3,4]. Recent evidence also suggests that dynamic epigenetic changes influence patterns of expression and play a key role in neurodevelopment[5]. Indeed, more than 100 modifications have been identified in natural cellular RNAs, including mRNAs, tRNAs, rRNAs, small nuclear RNAs (snRNAs), and small nucleolar RNAs (snoRNAs)[6]. Some of these modifications have a strong impact on gene expression, similar to dynamically regulated DNA and protein modifications[1,7].
TheN6-methyladenosine (m6A) modification is the most pervasive and important methylation observed in mRNAs[8,9]. In 1974, m6A was accidentally discovered in mRNA-enriched RNA fractions[10]. However, due to a lack of methods to detect the m6A site in mRNA at that time, interest in m6A gradually declined until proper methods for detecting were developed in recent years. Since then, research has suggested that m6A is closely related to many biological processes and transitions, such as RNA splicing, RNA stability, translation, mRNA nuclear export, and so on[8,11-13].
In this review, we summarize recent prominent progress in m6A modifications in neurodevelopment. This review is separated into three parts, including: (1) Writers, erasers, and readers that are involved in m6A methylation; (2) Methods of detecting and analyzing m6A modification; and (3) Recent publications on the important biological effects of m6A on the development of the nervous system. m6A RNA methylation has emerged as a new frontier in neuroscience, and this review provides insights into the mechanisms through which m6A modifications control expression.
1 Writers, erasers and readers of m6A
The m6A modification process is reversible and involves methyltransferase, demethylase, and RNA binding proteins. Among these, the key m6A methyltransferases, dubbed “writers”, include methyltransferase-like 3 (METTL3), METTL14, Wilms tumor 1-associating protein (WTAP), KIAA1429, RNA binding motif protein 15 (RBM15/15B), zincnger CCCH-type containing 13 (ZC3H13), METTL16, and zinc finger CCHC-type containing 4 (ZCCHC4). m6A demethylases (erasers) include fat mass and obesity-associated protein (FTO) and Alkb Homolog 5 (ALKBH5). m6A binding proteins (readers) include the YTH domain proteins, eukaryotic initiation factor 3 (EIF3), Insulin-like growth factor 2 mRNA binding proteins (IGF2BP), Proline rich coiled-coil 2 (PRRC2A), heterogeneous nuclear ribonucleoprotein C (HNRNPC), heterogeneous nuclear ribonucleoprotein (HNRNPG), heterogeneous nuclear ribonucleoprotein A2/B1 (HNRNPA2B1) and fragile X mental retardation protein (FMRP) (Fig.1).

Fig.1 N6-methyladenosine methylation and demethylation reactions Methyltransferase (m6A writer complex) containing METTL3, METTL14, WTAP, RBM15/15B, ZC3H13, KIAA1429, ZCCHC4 and METTL16 catalyzes m6A methylation, whereas the demethylases (m6A eraser) composed of FTO and ALKBH5 catalyze demethylation of m6A. The writers and erasers are localized primarily in the nucleus. A set of m6A-binding proteins (m6A reader) including YTHDC1, YTHDC2 and HNRNPC, HNRNPG, HNRNPA2B1 in nucleus and YTHDF1, YTHDF2, YTHDF3, EIF3, IGF2BP, PRRC2A and FMRP in cytosol determine the fate of target m6A-modified mRNA transcripts
1.1 m6A methyltransferase complex (“writers”)
The process of methylation of adenosine residues in eukaryotic mRNA to form m6A is catalyzed by methyltransferase complex that consists of multiple components, including METTL3, METTL14, WTAP, KIAA1429/VIRMA.
1.1.1 METTL3 In 1997, Rottman and colleagues isolated a 70 kD fraction that had S-adenosylmethionine (SAM)-binding activity (through column chromatography). It was originally identified as a methyltransferase component and was designated as MT-A70, but is now referred to as METTL3[14]. It is a highly conserved member of the SAM-dependent methyltransferase complex in mammals[15]. The METTL3 protein contains two conserved domains: the DPPW (Asp-Pro-Pro-Trp)-containing catalysing domain and the SAM-binding domain[14,16]. METTL3 is the major m6A-forming enzyme in polyadenylated mRNA.
1.1.2 METTL14 METTL14 is another component of the m6A methyltransferase complex and was identified through analysis of the human genome[17-19]. METTL14 is highly similar to METTL3 and also contains two conserved domains[16,19]. METTL3 and METTL14 form stable heterodimers with a stoichiometric ratio of 1∶1 in mammals[16]. The methyltransferase complex catalyzes methyl transfer in cellular m6A deposition on nuclear RNAs[20]. In addition, the combined methyltransferase activity of both enzymes is greater than that of either individual one alone. METTL3 is primarily considered the catalytic core, and METTL14 is the RNA-binding substrate[19,21].
1.1.3 WTAP Frayetal.have previously shown that WTAP is another important component of the m6A methyltransferase complex[22,23]. WTAP is a nuclear protein that plays a role in transcriptional and post-transcriptional regulation. WTAP has no catalytic activity related to m6A modification and has no effects on the activity of the METTL3-METTL14 complex. However, WTAP plays a key role in helping localize METTL3 and its binding partner, METTL14, in nuclear speckles to efficiently methylate mRNA[8,17,24]. Therefore, WTAP is a key adaptor of METTL3[8]. One prior study found that WTAP is a regulatory subunit necessary for the formation of m6A methyltransferase complexes (including others beyond METTL3 and METTL14) and plays an important role in gene expression and transcriptional regulation[24].
1.1.4 KIAA1429/VIRMA KIAA1429/VIRMA (also known as virus-like m6A methyltransferase-related protein) is a newly identified component of the RNA m6A methyltransferase complex that helps guide region-selective m6A deposition and regulates methylation[25]. KIAA1429 has been associated with the methylation complex in both proteomic screening and cellular studies[26-28]. Evidence indicated that the interactions between KIAA1429 and other methyltransferases may be important. Similar to WTAP, KIAA1429 localizes in the nuclear speckles in human cells[29]. Yue and colleagues reported that VIRMA/KIAA1429 recruits the catalytic core components (METTL3/METTL14/WTAP) to guide region-selective m6A methylation[30]. However, how KIAA1429 affects the methylated complex and why it is important for m6A remains to be determined.
1.1.5 RBM15/15B Prior work has showed that RBM15 and its paralog RBM15B are components of the m6A methyltransferase complex. Initially, RBM15 and RBM15B were shown to interact with WTAP through proteomic studies and co-immunoprecipitation analyses[29,31,32]. RBM15/15B is regarded as a mediator of methylation specificity[8,15]. The formation of m6A in X-inactive specific transcript (XIST) and mRNAs is mediated by the m6A methylated complexes RBM15 and RBM15B and in the mRNA transcriptome, where RBM15/15B is adjacent to the methylated DRACH sequence, but not to non-methylated DRACH sequences. RBM15 and RBM15B can recruit WTAP/METTL3 complexes to methylate nearby DRACH motifs to enhance m6A methylation[31].
1.1.6 ZC3H13 Several studies have demonstrated that ZC3H13 is another component of the m6A methyltransferase complex[33]. ZC3H13 is a zinc finger protein that plays an important role in regulating the methylation of RNA m6A in the nucleus. In mouse embryonic stem cells (mESCs), knockout of theZC3H13-coding gene significantly reduces the overall m6A levels of mRNA, impairs self-renewal, and triggers mESCs differentiation. In mESCs, ZC3H13 is necessary for the nuclear localization of the ZC3H13-WTAP-virilizer-hakai complex and plays an important role in the methylation of RNA m6A[34].
1.1.7 METTL16 The METTL3 homolog METTL16 (also known as METT10D) has been identified as an RNA m6A methyltransferase that can methylate both coding and non-coding RNA. Biochemical fractionation studies have suggested that most METTL16 protein is located in the cytoplasm. Of interest, METTL16 may exhibit different RNA binding preferences depending on its cell location[35]. METTL16 is an m6A methyltransferase of the mRNA precursor that maintains SAM homeostasis. It was found that METTL16 is an m6A-forming enzyme in mRNA and an m6A methyltransferase that targets U6 snRNA and MAT2A mRNA[8,36]. Whereas METTL16 is more likely to contain RNA substrates with double-stranded RNA adjacent to the consensus sequence. The METTL3/METTL14 complex does not require such a structural background. There are differences between METTL16 and METTL3/METTL14 in terms of mechanisms for identification and regulation of substrates[36].
1.1.8 ZCCHC4 28S rRNA carries a single m6A modification and the site was found at position A4220 within a stem-loop structure. Pintoetal.found that ZCCHC4 is a methyltransferase responsible for the introduction of m6A at position 4 220 of 28S rRNA[37]. ZCCHC4, a 28S rRNA-specific m6A methyltransferase, binds to S-adenosyl-L-homocysteine[20]. ZCCHC4 is located in ribosome-assembled nucleoli, and proteins involved in RNA metabolism are over-represented in ZCCHC4-interacting proteins, suggesting that ZCCHC4 may play a key role in mRNA translation[37]. Prior studies have also shown that ZCCHC4 interacts with many RNA-binding proteins.
In short, the molecules METTL3, METTL14, and WTAP jointly constitute the methyltransferase complex, with METTL3 serving as the catalytic core and METTL14 as the RNA binding substrate. WTAP is a subunit necessary for the formation of the complex. KIAA1429 recruits core components and directs methylation. RBM15 and RBM15B can recruit WTAP/METTL3 complexes to promote methylation. ZC3H13 and METTL16 are recently discovered methyltransferases that act synergistically with the core components to promote methylation (Fig.1).
1.2 m6A demethylases (“erasers”)
It has been speculated that m6A RNA methylation is a reversible process. Indeed, shortly following the discovery of methyltransferases, demethylases were gradually discovered.
1.2.1 FTO FTO was the first methyltransferase discovered. Jia and colleagues first showed that FTO can significantly catalyze m6A demethylation in mRNAinvitro[38,39]. siRNA knockdown of FTO resulted in increased levels of m6A in mRNA, while overexpression of FTO resulted in decreased levels of m6A in human cells. FTO is a member of the Alkb protein family and FTO also is associated with obesity[40]. Bioinformatic analysis has revealed that FTO catalyzes oxidative demethylation. m6A in polyadenylated RNA has been identified as a target of FTO[41,42]. However, Maueretal.proposed that FTO may be the eraser for N6, 2′-O-dimethyladenosine (m6Am), impacting mRNA stability[8,43]. FTO also can mediate the demethylation of m1A in tRNA in the nucleus and cytoplasm, and may regulate translation together with the demethylation of cytoplasmic cap m6Am mRNA[44].
1.2.2 ALKBH5 ALKBH5 is a member of the Alkb protein family. It is the second-identified m6A demethylase and displays m6A demethylation activity comparable to FTO. ALKBH5 localizes to the nucleus and plays a role in the regulation of the nuclear export of mRNA as well as the regulation of splicing and stability of mRNAs[45,46]. Further, ALKBH5 plays a key role in immune responses to viral infection[47]. Prior work has shown that deletion of ALKBH5 in glioblastoma stem-like cells results in a unique m6A modification pattern for specific transcripts, resulting in changes in gene expression[48].
In summary, FTO and ALKBH5 are the two demethylases that have been identified to date, and exhibit comparable m6A demethylation activity. FTO affects mRNA stability and alternative splicing[38,49], while ALKBH5 affects mRNA nucleus output and mRNA splicing (Fig.1).
1.3 RNA-binding proteins (“readers”)
m6A indirectly affects RNA processing via specific RNA-binding proteins. To date, several m6A-binding proteins have been identified from mammalian cellular extracts[16].
1.3.1 YTH domain proteins The YTH domain family includes both the cytoplasmic YTH domain-containing family (YTHDF1, YTHDF2, and YTHDF3) and nuclear m6A readers (YTHDC1 and YTHDC2)[50-52]. Rechavi and colleagues previously identified YTHDF2 and YTHDF3 as m6A-binding proteins. These proteins are named for their YTH domains that are responsible for removing transcripts of genes related to meiosis[50]. After the initial identification of YTHDF2 and YTHDF3 as m6A-binding proteins, several groups verified those reports using gel shift assays and crystallography[53]. These follow-up studies showed that the YTHDF1, YTHDF2 and YTHDF3 bind to all the m6A sites in mRNA, and the nucleus-enriched YTHDC1 binds to some of the m6A sites in mRNA as well as the m6A site in nuclear non-coding RNA using the iCLIP (individual-nucleotide-resolution UV crosslinking and immunoprecipitation) method[31,54]. YTHDC2 seems to function as an ATP-dependent RNA helicase[16,55]. Elongation-promoting effect of coding sequence (CDS) methylation requires the RNA helicase YTHDC2 containing m6A and RNA helicases containing YTHDC2 were recruited into CDS to participate in m6A methylation and promote the translation of structural mRNAs[56].
1.3.2 EIF3 mRNA modified with m6A can be translated by EIF3, which has been confirmed through the use of cell lysates lacking EIF3. About 35% of the m6A sites in the 5′-UTR overlap with the EIF3 splice site, supporting the physiological significance of EIF3 as an m6A-binding protein complex. Therefore, m6A modification is an important mechanism by which EIF3 is recruited into mRNA, and EIF3 can bind directly to the m6A sites in the 5′-UTR of mRNA to promote mRNA translation[57].
1.3.3 IGF2BP IGF2BPs, including IGF2BP1/2/3, are unique readers for the m6A, targeting thousands of mRNA transcripts by identifying consistent GG (m6A) C sequences. IGF2BPs promote stability and storage of target mRNA in an m6A-dependent manner under both normal and stress conditions, thereby affecting gene expression. IGF2BP can make the target gene and relevant translation more stable[9,57]. IGF2BP is of great importance in post-transcriptional gene regulation.
1.3.4 PRRC2A PRRC2A is a recently identified m6A reader that stabilizes mRNA translation by binding the GGACU motif consistent in the CDS region in an m6A-dependent manner[9,58]. PRRC2A plays an important role in oligodendrocyte specification by functioning as an m6A reader. It has been shown that PRRC2A was highly expressed in oligodendrocyte precursor cells. PRRC2A stabilizesOlig2 mRNA in an m6A-dependent manner by binding to theOlig2 coding sequence with a consistent GGACU motif and directly regulatesOlig2 expression bothinvivoandinvitro[58].
1.3.5 HNRNPC, HNRNPG and HNRNPA2B1 HNRNPC is an abundant nuclear RNA-binding protein. It binds the nascent RNA transcripts and affects the stability, splicing, export, and translation of the pre-mRNA[59-63]. HNRNPA2B1 and HNRNPG are also identified as nuclear readers of m6A and change alternative splicing patterns[64,65]. HNRNPA2B1 also binds primary miRNA transcripts and promotes primary miRNA processing[8,66].
1.3.6 FMRP FMRP is encoded by the fragile X mental retardation gene (FMR1) and mutation will result in fragile X syndrome, one of the causes of intellectual disability[67]. FMRP is a selective RNA-binding protein and also is a reader of m6A to promote nuclear export of methylated mRNA targets[11,68]. It also plays roles in mRNA localization[11,68].
Briefly, among the YTH domain proteins, YTHDF1, YTHDF3, and YTHDC2 may be related to translation processes, while YTHDF2 are related to mRNA stability and delay. YTHDC1 is related to nuclear export and mRNA splicing. FMRP also plays roles in mRNA localization and nuclear export. EIF3 binds to m6A-containing mRNA to regulate mRNA translation. IGF2BPs and PRRC2A promote the stabilization and storage of mRNA in an m6A-dependent manner. HNRNPC is responsible for pre-mRNA processing, while HNRNPA2B1 and HNRNPG promotes primary miRNA processing (Fig.1).
2 m6A mapping approaches
RNA m6A was first discovered in the 1970s. However, due to the lack of methods to detect the m6A site precisely, activity research gradually decreased. In recent years, researchers have identified several methods to detect the m6A site. With the emergence of high-throughput sequencing technology, mass spectrometry, and other methods, Rechavi’s research group developedN6-methyladenosine-sequencing (m6A-seq) by combining co-immunoprecipitation with high-throughput sequencing technology. Since then, interest in m6A has been renewed, with increased publication activity related to m6A (Fig.3B). The current technical methods for m6A detection include high-throughput sequencing, colorimetry, and liquid chromatography-mass spectrometry (LC-MS). Specific methods include m6A-seq, m6A individual-nucleotide-resolution crosslinking and immunoprecipitation (miCLIP-seq), LC-MS/MS, and so on. Here we will take a closer look at these emerging technologies.
m6A-seq involves in use of highly specific anti-m6A antibodies to isolate immunoprecipitated methylated RNA fragments from a random fragment transcriptome. Then, these fragments are sorted in parallel and the signal enrichment position relative to the input control is determined (Fig.2A)[7].

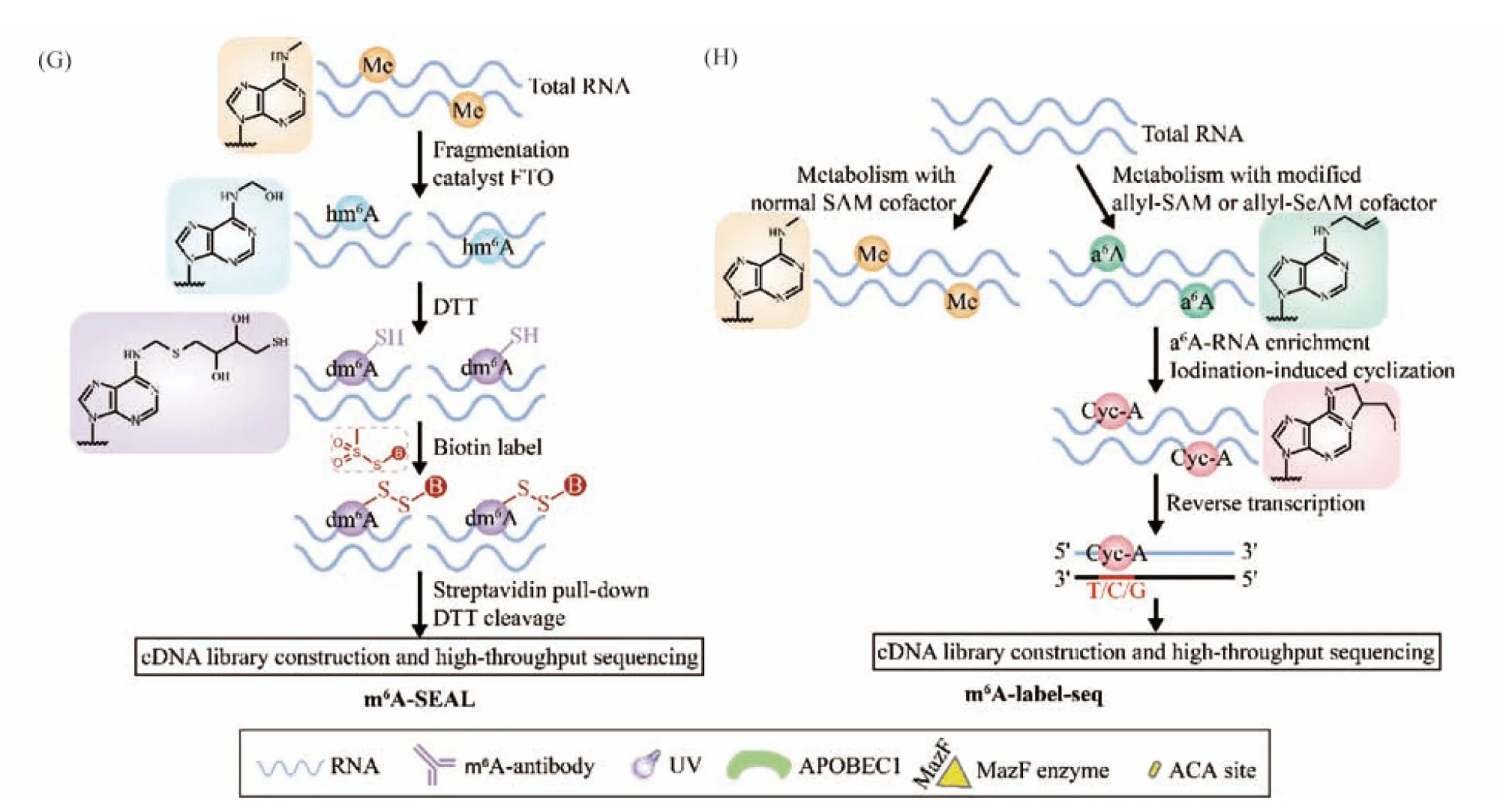
Fig.2 Schematic diagram of seven methods for transcriptome-wide mapping of adenosine methylation (A) m6A-seq: total RNA is removed rRNA and is chemical fragmented into ~150-nt-long oligonucleotides. Fragmented RNA is subjected to immunoprecipitation using an m6A-specific antibody and IgG antibody. Eluted m6A-containing fragments (IP), IgG-containing fragments (IgG control) and untreated fragments (Input control) are converted to cDNA and followed by Illumina sequencing. (B) PA-m6A-seq: 4SUs are integrated into RNA, after the full-length mRNA is IP, the sample is irradiated with 365 nm ultraviolet light, triggering cross-linking, the cross-linked RNA is digested to about 30 nt with RNase T1. Then total RNA is extracted with TRIzol reagent and reverse transcribed. (C) miCLIPy-seq: The fragmented RNA was obtained in the same way as (A) and incubated with m6A antibody. Through cross-linking with UV light (254 nm), antibody-RNA complexes are recovered. Then RNA is released by proteinase K and reverse transcribed. The exact location of m6A could be found in terms of the cross-link-induced mutations (C-T transition) and truncations during reverse transcription. (D) DART-seq: APOBEC1 is fused into the YTH domain to trigger the C-U deamination at cytidine residues near the m6A site. Then total RNA is isolated and reverse transcribed. C-to-U mutations are detected in RNA-seq to identify sites of m6A. (E) m6A-REF-seq: mRNA is digested into fragments by MazF. Then fragments are converted to cDNA and followed by Illumina sequencing. A parallel sample batch demethylated by FTO in advance as the negative control is included and only sites with reduced methylation ratios in the FTO-treated group were considered to be accurate m6A sites. (F) MAZTER-seq: mRNA is digested into fragments by MazF. After end repair and ligation of an adaptor, fragments are reverse transcribed and amplified by polymerase chain reaction (PCR). (G) m6A-SEAL: RNA demethylase FTO oxidizes m6A to hm6A and hm6A can be further converted to dm6A with a DTT-mediated thiol-addition reaction. The free sulfhydryl on dm6A is marked with the mRNA-biotin and could be captured by streptomycin avidin. The RNA is further fragmented by DTT and RNA fragments containing m6A modifications were enriched for subsequent high-throughput sequencing. (H) m6A-label-seq: RNA allyl-modified methionine was introduced and generates cofactor allyl-SAM or allyl-SeAM through the action of intracellular MAT. Then RNA could be metabolically modified with a6A at the supposed m6A-generating adenosine sites. a6A-RNA is enriched and converted to Cyc-A through iodine-induced cyclization. Then reverse transcriptase is used to construct cDNA library for high-throughput sequencing and the m6A site could be identified by analyzing the mutation site
Since the methods for locating m6A sites are still limited, Heetalproposed photo-crosslinking-assisted m6A-sequencing (PA-m6A-Seq) on the basis of Photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP). The photoactivatable ribonucleoside, 4-thiouridine (4SU) or 6-thioguanosine (6SG) is incorporated into mRNA. Under UV irradiation at 365 nm, RNA-binding proteins were covalently cross-linked[69].
Since the immunoprecipitated RNA fragment may contain m6A at any point in its length, multiple different fragments containing m6A may be overlapped, modified residues cannot be identified at specific loci, and peak calling in bioinformatics may cause some bias. Therefore, Linder developed another single-nucleotide-resolution m6A mapping technique called miCLIP[70]. After ultraviolet light-induced antibody-RNA cross-linking and reverse transcription, m6A antibodies can induce specific mutational signatures at m6A residues. In miCLIP, the shape of the peak does not affect the recognition of m6A[70](Fig.2C). Later, Heetaldeveloped m6A crosslinking immunoprecipitation sequencing (m6A-CLIP-seq). With only approximately 1 μg of mRNA as starting material, this approach can profile m6A at a resolution of 100 nucleotides. m6A-CLIP-seq uses 254 nm UV irradiation and less starting material and provides profiles with greater resolution. In addition, m6A-CLIP-seq can extract RNA from freshly separated tissue samples[71]. This approach optimizes miCLIP and requires less RNA initiation.
Meyeretal. developed an antibody-free method for detecting the m6A site called DART-seq. In DART-seq, the cytosine deaminase Apolipoprotein B MRNA Editing Enzyme Catalytic Subunit 1 (APOBEC1) is fused with the YTH region that binds to m6A. APOBEC1-YTH expression in cells induces C to U deamination at sites near the m6A residues and can be detected by standard RNA-seq. With this method, one can detect m6A accumulation and analyze m6A distribution along the length of individual transcripts (Fig.2D)[72].
Based on the recognition of the ACA motif, Zhang and colleagues[73]developed an accurate, high-throughput, antibody-independent m6A recognition method called m6A-REF-seq (m6A-sensitive RNA-Endoribonuclease-Facilitated sequencing). They used independent methods to validate methylation status and abundance of individual m6A site. They applied this method to 5 tissues from human, mouse, and rat, and found that the m6A site was mononucleotide specific. Further, m6A sites tend to cluster at homologous sites across different species (Fig.2E)[73].
Garcia-Campos developed RNA digestion via m6A sensitive RNase (MAZTER-seq) to enable systematic quantitative profiling of m6A at a single-nucleotide resolution at expressed sites, building on differential cleavage by an RNase. MAZTER-seq is based on an RNA enzyme MazF which can specifically cut the non-methylated ACA site from the beginning. The MazF-treated mRNA fragments are reversely transcribed, and sequenced. Methylated ACA sites are then identified and quantified by MAZTER-MINE, a computational pipeline that quantifies the number of times each transcribed ACA site is started, finished, and read[74]. MAZTER-seq allows quantitative studies of the regulation of m6A in subcellular parts, different cell
types, and disease states. The advantage of this method is that it enables analysis of the m6A code, exploration of its function, and tracking of the dynamic response and genetic perturbation of methylation levels (Fig.2F)[74]. One of the limitations of this method is that it can only recognize the methylation of ACA sites, and ACA only accounts for about 16% of m6A motif. However, the publication of this method has promoted the development of the research field of m6A and provided new ideas for the study of m6A.
Recently, Jia group developed m6A-SEAL, an elegant FTO-assisted m6A selective chemical labeling method that uses the demethylase FTO as a catalyst to convert the chemically inert m6A on mRNA into N6 hydroxymethyl adenine (hm6A) and then uses the thiol group of dithiothreitol (DTT) to react with hm6A to convert the unstable hm6A into a more stable thiol addition product, N6-dithiolsitolmethyladenosine (dm6A). The free sulfhydryl on dm6A reacts rapidly with methane thiosulfonate (MTSEA) to mark the position of m6A on the mRNA-biotin, which can be captured by streptomycin avidin. Using this method, enriched RNA fragments containing m6A modifications can be captured for subsequent high-throughput sequencing (Fig.2G)[3].
At around the same time, Liu’s research group reported a high-throughput sequencing method called m6A-label-seq, which introduces an allyl-modified methionine and generates cofactorS-adenosyl methionine (allyl-SAM) orSe-allyl-L-selenohomocysteine (allyl-SeAM) through the action of intracellular methionine adenosyl transferase (MAT). Then, the m6A sites are modified to form N6-allyladenosine (a6A) and Cyc-A is generated after the addition of iodine. Base mismatch occurs when this RNA Cyc-A is reverse transcribed into complementary DNA (cDNA). Finally, a single-base resolution map of the full transcriptome m6A site can be obtained through high-throughput sequencing and bioinformatics analysis (Fig.2H)[54,75].
Research interest in m6A continues to rise, with increasing number of publications related to m6A methylation detection methods in recent years (Fig.3A). Among these methods, m6A-seq is most widely used, possibly because this was the first method developed and enables identification of m6A at the genome-wide or transcriptional level. This method is convenient, fast, and cheap and can be used to conduct a qualitative analysis of the mRNA regions with hypermethylation. However, it can only identify the hypermethylated regions of m6A and cannot achieve the resolution of a single base. Subsequently, the emergence of PA-m6A-Seq improved the resolution and allowed the precise location of the sites where m6A modification occurred. The appearance of miCLIP enabled single-base resolution of m6A and overcome the distance limit. Other methods are not yet widely used (Fig.3A), but they do have their advantages. DART-seq can detect the accumulation of m6A by standard RNA-seq and analyze the distribution of m6A along a single transcribed length. m6A-REF-seq verifies the methylation status and abundance of a single m6A site using an independent method. m6A-CLIP -seq uses less starting material to achieve greater and higher resolution. MAZTER-seq system quantitatively analyzes the resolution of single nucleotide at the expression site of m6A. m6A-SEAL has high sensitivity and specificity and can be used in a small number of mRNA samples. m6A-label-seq has higher accuracy and is suitable for the identification of various m6A motifs of intracellular RNA, which provides a strong basis for future research on m6A methylation sites and state evolution of a wide variety of RNAs. These methods are each suitable for different situations in practical applications and should be chosen in line with research needs and experimental conditions. In the future, researchers will likely optimize and modify these methods to achieve single-base resolution and imaging of m6A.
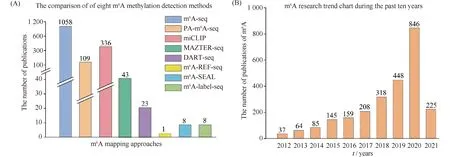
Fig.3 Research trends of m6A (A) The comparison of the number of publications in eight methods for transcriptome-wide mapping of adenosine methylation. The bar chart shows the number of publications of these methods which were referenced in the database. (B) The publication of m6A shows an obvious rising trend. The bar chart shows the number of publications of m6A in the database
In the study of m6A, in addition to the continuous improvements and innovation in experimental methods, many scholars have also developed softwares based on different mathematical models to further predict the m6A site in RNA and further annotate the function of m6A methylation. Among these, iRNA-Methyl[76], SRAMP[77], M6AMRFS[78], M6APred-EL[79], and DeepM6ASeq[80]are used to predict loci, and m6Acomet[81]was used to annotate m6A. As the methods are constantly updated, researchers have found that RRACH (R = G or A; H = A, C or U) is a motif closely related to m6A modification[6]. The modification of m6A mainly occurs in the CDS, near the stop codon and 3′-untranslated regions (3′ UTR) region[12].
3 The role of mRNA m6A methylation in neurodevelopment
mRNA methylation is an important epigenetic modification. Many studies have found that m6A is highly expressed in the brain, and m6A methylation plays an indispensable role in the nervous system in the process of neurodevelopment, including neurogenesis, axonal growth, neural differentiation, and synapse formation. Next, we will review the effects of m6A on different aspects of neurodevelopment.
3.1 m6A in neurogenesis
Neurogenesis is the process by which neurons are produced from different types of neural progenitor cells (NPCs, including neural stem cells)[82]. During development, neurogenesis results in a large diversity of neurons in the nervous system. Proper maintenance of neurogenesis is essential for normal neurodevelopment, and interference with neural stem cells self-renewal and neurogenesis during development can directly lead to neurological and psychiatric disorders. Studies have shown that m6A regulates neurogenesis[83].
Yoonetal. analyzed the effect of m6A signaling pathways on cortical neurogenesis using an embryonicMettl14 gene knockout model. They found that the radial glial cell cycle was prolonged and cortical neurogenesis extended to the postnatal stage in this model[71]. In addition, m6A was found to be associated with mRNA turnover, and the m6A marker promoted mRNA decay[1,84].
Caoetal. found that specific ablation of FTO inhibits neurogenesis and neuronal development in adults, and that FTO regulates adult neural stem cells through thePdgfra/Socs5-Stat3 pathway[85]. FTO is expressed in adult neural stem cells and neurons, and the loss of FTO reduces brain size and weight. FTO also reduces proliferation and neuronal differentiation of adult neural stem cellsinvivo, and changes the expression of key components of m6A-labeled brain-derived neurotrophic factor pathway, leading to impaired learning and memory[86].
Loss of METTL3 not only inhibits the development of neurons and makes the differentiation of adult neural stem cells more likely to be of glial lineage, but also affects the morphological maturity of newborn neurons in the adult brain[87]. m6A-seq showed that m6A was mainly enriched in transcripts related to neurogenesis and neuronal development. There is m6A on the transcript of histone methyltransferase EZH2, which reduces the expression of EZH2 protein and the corresponding H3K27 me3 level after the knockout of METTL3. Overexpression of EZH2 can rescue neurogenesis and neuronal development defects caused by loss of METTL3[87]. METTL3 likely plays a role in profilin regulation in neurogenesis[88].
The Nestin-Cre-mediated knockout ofPrrc2ain mice led to significant hypomyelination as well as shortened lifespan, motor impairment, and cognitive deficits, suggesting thatPrrc2ais involved in oligodendrocyte progenitor cell (OPCs) proliferation and oligodendrocyte fate determination[89]. Degradation of m6A-modified transcripts may help maintain proper cortical neurogenesis. Abnormal m6A can affect time specification and cell cycle progression of neural progenitor cells[2].
In summary, the m6A "writer" METTL14 and "reader" PRRC2A are closely related to the cycle of neurogenesis, and their absence can significantly prolong the neurogenesis cycle. The m6A "eraser" FTO may influence neurogenesis by impacting the differentiation process of neural stem cells.
3.2 m6A in neural differentiation
Loss of YTHDF2, an m6A reading protein in mouse, results in impaired embryonic neural development, affecting the self-renewal of nerve stem cells and the spatiotemporal generation of neurons and other types of cells. YTHDF2 regulates neural development by promoting m6A-dependent degradation of neural development-related mRNA targets[90]. Through m6A-seq, researchers found a set of m6A-modified transcripts directly regulated by YTHDF2 and related to neurodevelopment[91]. The absence of YTHDF2 in induced pluripotent neural stem cells leads to increased stability of gene transcripts, loss of pluripotency, and induction of neuron-specific gene expression. YTHDF2 inhibits translation of neuron-specific mRNAs in induced pluripotent stem cells and promotes its rapid and coordinated upregulation during nerve induction[92].
Edensetal. found that FMRP is an m6A reader that promotes the nuclear export of methylated mRNA during neural differentiation. The absence of either FMRP or m6A methyltransferase METTL14 leads to nuclear accumulation of mRNAs related to neural differentiation, resulting in delayed differentiation of neural progenitor cells in mice[11]. InFmr1 (gene encoding FMRP protein) knockout (KO) mice andMettl14 conditional knockout (cKO) mice, cell cycle progression is delayed and the excitation-inhibition balance of the neural circuits is disrupted. BothMettl14 cKO andFmr1 KO result in nuclear retention of m6A-modified FMRP target mRNA that regulates neural differentiation, thereby affecting neural differentiation[11].
An analysis has demonstrated that the development of oligodendrocyte lineage is accompanied by dynamic changes in m6A transcription[92]. Inactivation of METTL14 leads to a decrease in oligodendrocytes and the hypomyelination of the central nervous system. Ablation of METTL14 invitrodestroys the maturation of oligodendrocytes after mitosis and has a significant effect on the transcriptome of OPC and oligodendrocytes. Loss of METTL14 leads to abnormal splicing of numerous RNA transcripts[92].
The size and division rate of drosophila neural stem cells (neuroblasts) are controlled by the highly conserved RNA binding protein Imp (IGF2BP) through one of its most important binding targets in the brain,mycmRNA. One study found that Imp stabilizesmycmRNA, leading to an increase in MYC protein levels, an increase in the size of nerve cells, and faster division[93]. Across the entire development process, the decline in Imp level limits the stability ofmycmRNA, thus inhibiting the growth and division of neuroblasts, and the expression of heterogeneous Imp is related to the stability ofmycmRNA between individual neuroblasts in the brain[93].
Briefly, the m6A "reader" YTHDF2 is closely related to self-renewal of neural stem cells and generation of neurons and other types of cells. The m6A "reader" IGF2BP promotes growth and division of nerve cells by stabilizingmycmRNA. FMRP protein promotes nuclear transport of m6A methylated mRNA and promotes differentiation of neural progenitor cells. Loss of m6A "writer" METTL14 affects myelination in the central nervous system, further affecting the process of neuronal differentiation.
3.3 m6A in axon guidance, synaptic formation and plasticity
Synapses connect billions of neurons to process information and drive behaviors[94]. Neuronal communication is a dynamic process resulting from the integration of synaptic elements at the intramolecular, intermolecular, and subcellular scales[95]. Adequate synaptic function is a necessary prerequisite for all neural processing, including higher cognitive functions such as learning and memory[96].
Specific ablation of YTHDF1 in the spinal cord disturbed the pre-crossing axon guidance[97]. YTHDF1 promotes axon guidance receptor Robo3.1 translation, and YTHDF1 cKO mice are deficient in Robo3.1 expression and cross-axonal pre-exploration. This suggests that m6A modification and YTHDF1 regulate Robo3.1 translation and further control axon guidance[97].
By knocking down YTHDF3 in hippocampal neurons to block m6A-mediated regulation, the expression of a microtubule plus-end tracking protein APC is altered[94]. Merkurjevetal. found that altered APC expression may contribute to the synaptic dysfunction including immature spinal morphology and inhibition of excitatory synaptic transmission which accompanied by a reduction in postsynaptic density-95 (PSD-95) clusters and a reduction in the surface expression of AMPA receptor subunit GluA1[94]. These synaptic abnormalities may cause synaptic plasticity abnormality, disrupt learning and memory, and cause neurodevelopmental disorders[94].
FTO is enriched in axons and can be locally translated. Knockdown of FTO in axons leads to increases in m6A levels and decreased translation of GAP-43 mRNA[98]. Downregulation of Taurine Up-Regulated 1(TUG1) promotes axonal development in cultured hippocampal neurons, while overexpression of TUG1 leads to axonal growth defects during embryonic cortical development[98]. Guoetal. found that knockout of TUG1 rescued axonal development defects in FMRP deficient neurons[99]. The m6A modification plays a key role in synaptic regeneration of neurons in mature mice[100]. An increase in m6A in neurons alters the transcriptome response to synaptic plasticity[101].
In conclusion, m6A "reader" YTHDF1 and "eraser" FTO are related to axon formation, growth, and development, and their loss may cause axon growth defects. Further, the m6A "writer" METTL3 is closely related to the expression and function of synapses.
3.4 m6A in the cerebellar development
Several reports have analyzed the expression of m6A writers METTL3, METTL14, and WTAP, erasers ALKBH5 and FTO in the cerebellum of mice. Abnormal expression of METTL3 and ALKBH5 leads to an unbalance in RNA methylation, which leads to cerebellar development defects[102]. This suggests that knockout of theMettl3 gene had negative effects on Purkinje cell number, lamellar structure, dendritic formation, and organization of glial cell fibers in the cerebellum[102].
4 Problems and prospects
Discovery of m6A in 1974 suggested that m6A was a broadly prevalent modification that played a role in the regulation of gene expression. More recently, with the emergence of innovative scientific methods, the ability to identify and characterize m6A has been improved, furthering our understanding of this modification. These studies have revealed that m6A plays a crucial role in gene expression regulation. In this paper, we introduce the molecules involved in the methylation process of m6A in detail, and summarize eight existing methods to detect the m6A site. Each method has its own merits and one can choose the appropriate method according to the research needs. We also review the effects of m6A on neurodevelopment in recent years.
In summary, mRNA methylation is an important epigenetic modification and m6A is highly prevalent in the brain. m6A has a wide range of effects on neural development, including neurogenesis, neuron differentiation, synaptic abnormity and so on. However, m6A is a relatively new field of study and many questions remain unknown. Although changes in epigenetic modification of mRNA may have an impact on neurodevelopment, these findings need to be confirmed in future studies. m6A modification is not an isolated event, but rather interacts with other mRNA modifications to produce certain cellular characteristics during neural differentiation and subsequent maturation. Emerging long-read sequencing technologies such as Nanopore and PacBio will provide us with further insights into m6A. In addition, the advent of single-cell sequencing will broaden our understanding of the differences between different nerve cell types and the specific effects of m6A on the development of specific neural cell types.
