
NH3分子在Fe原子掺杂石墨炔表面的吸附特性研究
2022-09-01 01:09杨文涛王建军杨林峰
中原工学院学报 2022年3期
杨文涛, 赵 宾, 王建军, 杨林峰
(中原工学院 理学院, 河南 郑州 450007)
随着全球人口增加以及经济的快速发展,大气污染问题日益突出,对大气环境的检测越来越重要。环境测量站、气象监测站对空气质量的检测离不开相应的气体检测传感器。基于此需求,许多科研人员致力于有毒、有害化学气体连续检测所用高灵敏度气体传感材料的研发[1]。众所周知,氨气(NH3)是一种低沸点化合物,具有很强的挥发性。因此,对氨气的有效检测和抑制已成为目前大气环境检测和控制工作的重中之重[2]。
2010年,中国科学院研究人员让六炔基苯在铜片的催化作用下发生偶联反应而成功制备石墨炔(Graphyne,GY)以来,GY吸引了物理、化学、能源等领域的众多研究者,目前已在多个领域展现出良好的应用前景[3-4]。GY作为一种新型的二维(2D)碳同素异形体,具有良好的化学稳定性、较大的比表面积和丰富的不饱和键,是优良的半导体材料[5-6]。GY中的碳原子以sp、sp2和sp33种杂化状态存在,从而导致了石墨炔的多样性,目前研究者关注较多的α-石墨炔、β-石墨炔、γ-石墨炔、6,6,12-石墨炔等[7-8],有望在纳米电子和光电子器件、储能、化学传感器、自旋电子学、电池电极等领域得到广泛的应用[9-12]。对气敏性传感器、锂离子电池、氧化反应以及还原反应的研究,无不涉及单原子、气体分子或者原子团簇在材料表面的吸附性,因此探究材料表面的吸附特性是非常重要的[13-14]。对气敏性传感器来说,其正常工作必然要求材料表面对气体分子具有较高的灵敏度,气体分子被吸附后能够引起传感系统电子性质的改变[15-17]。掺杂方法常用于调控2D材料的电子特性和其他物理性质,受到了科研人员的青睐。在纳米结构中掺杂原子、分子或分子基团,是一种可行的调控材料性能的方法[18]。为了论证GY作为传感材料的可行性,本文对过渡金属Fe原子掺杂GY表面(GY-Fe)与NH3分子之间的相互作用进行探讨,对比本征GY和GY-Fe表面吸附NH3分子的机制,研究NH3分子在Fe原子掺杂石墨炔表面的吸附特性。
1 基于第一性原理的计算模型
EB=Etotal-Egraphyne-EFe
(1)
式中:Etotal为Fe原子嵌入GY体系的总能量;Egraphyne为本征GY的能量;EFe为孤立Fe原子的能量。结合能EB的绝对值越大,说明GY-Fe体系越稳定。
为探究NH3分子与不同衬底的相互作用,分别计算了气体分子NH3在本征GY和GY-Fe表面的吸附能,并对不同的吸附角度和吸附位置进行区分,以便找到气体分子NH3在不同衬底上最稳定的吸附构型。气体分子NH3与Fe原子掺杂GY表面的吸附能为:
(2)

2 Fe嵌入单层GY的结构与稳定性
研究NH3分子在GY-Fe表面的吸附性能之前,本文对GY-Fe的几何结构进行优化,得到了图1所示GY-Fe的稳定构型。
图1(a)中,Fe原子嵌入GY表面的C1位置;结构优化显示,Fe原子与其周围3个C原子形成了Fe-C键。图1(b)中,Fe原子嵌入GY表面的C2位置;结构优化显示,Fe原子与其周围3个C原子也形成了Fe-C键,且Fe原子移动到了炔环中心附近。值得注意的是,GY-Fe的结构表面没有凸起或者凹陷,也就是说Fe原子的掺杂并没有改变GY的二维平面结构。计算可得表1所示GY-Fe的结构参数。结构参数包括:Fe原子与GY表面的结合能EB、Fe原子和C原子之间的键长dFe-C、Fe原子与GY表面之间的电荷转移量ΔQ和磁矩M。

表1 GY-Fe的结构参数Tab. 1 Structural parameters of GY-Fe
为研究掺杂Fe原子对单层GY电子结构的影响,分别计算了掺杂前后单层GY系统投影到Fe原子和相邻C原子轨道的电子分态密度(Partial Density of States,PDOS)。GY-Fe相电子分态密度与能量的关系如图2所示。

图2 GY-Fe相电子分态密度与能量的关系Fig. 2 The relationship between partial density of states and energy of GY-Fe
从图2可以看出,杂质Fe原子嵌入而诱导的缺陷态密度峰分布在费米能级周围,并且自旋向上的缺陷态密度峰呈现半占据态,导致密度峰的缺陷态主要来自于Fe原子3d轨道的贡献,同时,Fe原子3d轨道和相邻C原子2p轨道的电子态之间存在较强的杂化交叠,电子结构的变化也意味着Fe原子与相邻3个C原子之间的相互作用,会导致掺杂周围电荷的转移及重新分布。根据Bader电荷分析可知,对于掺杂C1和C2位置,约分别有0.825 e和0.740 e的电荷从杂质Fe原子上转移到了GY表面,且转移电荷主要由相邻的3个C原子接收。图3所示为GY-Fe的表面差分电荷密度。这里,电荷密度等值面的电荷密度为0.002 5 e/bohr3。
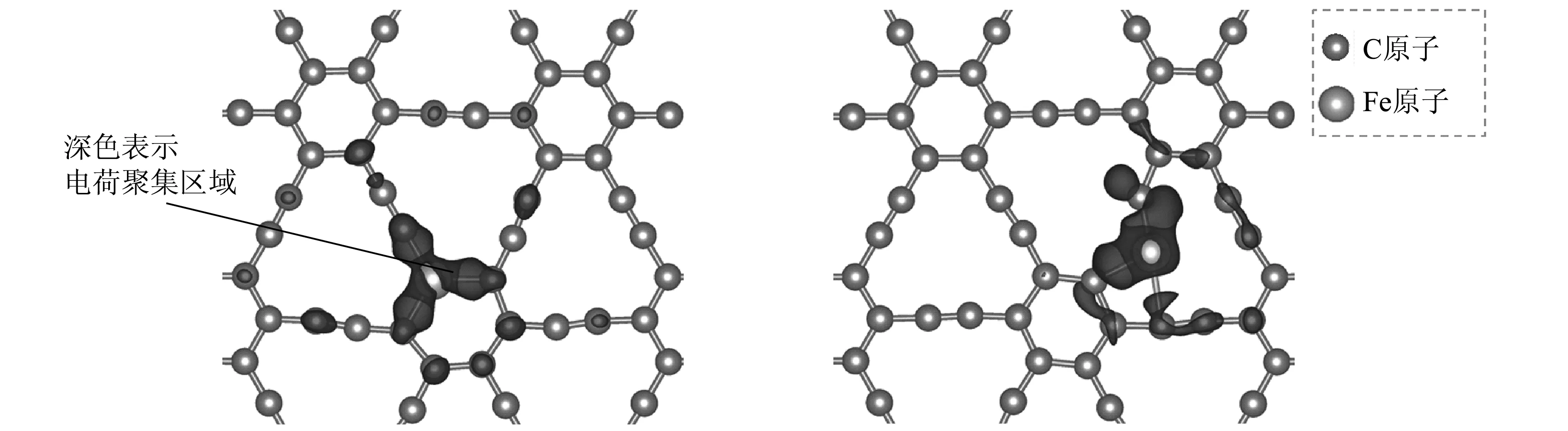
(a) C1位置 (b) C2位置 图3 GY-Fe的表面差分电荷密度Fig. 3 Charge density difference of GY-Fe surface
从图3看到了结构优化后的电荷转移及重新排布情况,即:优化后Fe原子与相邻C原子之间的作用,导致了掺杂周围电荷的转移及重新分布,且电荷主要聚集在Fe-C 键周围。这说明,掺杂系统形成了Fe-C化学键。
3 NH3分子在GY-Fe表面的吸附特性
为研究NH3分子在本征GY和GY-Fe表面的吸附特性,本文计算了NH3分子在本征GY和GY-Fe表面的吸附能Ea、NH3分子与不同衬底最近的距离d、NH3分子中N-H键长L、H-N-H键角θ、NH3分子与不同衬底之间的电荷转移量ΔQ′(见表2)。

表2 GY和GY-Fe表面吸附NH3分子的性质Tab. 2 Adsorption properties of NH3 molecule on the GY and GY-Fe surfaces

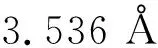

(a) C1位置俯视图 (b) C2位置俯视图

(c) C1位置侧视图 (d) C2位置侧视图 图4 NH3分子吸附在GY表面的稳定构型Fig. 4 Optimized structure of NH3 adsorbed at GY surface
Fe原子掺杂的GY表面对NH3分子的吸附情况又如何呢?结构优化后,NH3分子在GY-Fe表面形成了一个稳定的吸附构型(见图5)。
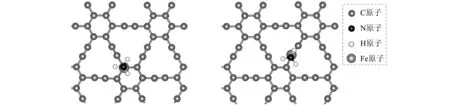
(a) C1位置俯视图 (b) C2位置俯视图
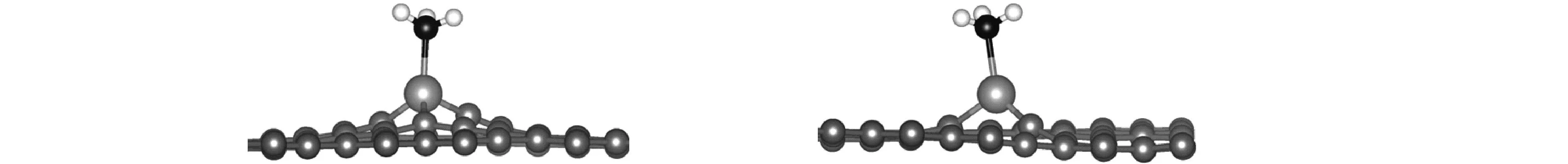
(c) C1位置侧视图 (d) C2位置侧视图 图5 NH3分子吸附在GY-Fe表面的稳定构型Fig. 5 Optimized structure of NH3 adsorbed at GY-Fe surface
为了更深入地了解NH3分子在GY-Fe表面的这种化学吸附行为,本文计算了GY-Fe-NH3系统的电子分态密度。GY-Fe-NH3系统电子分态密度与能量的关系如图6所示。
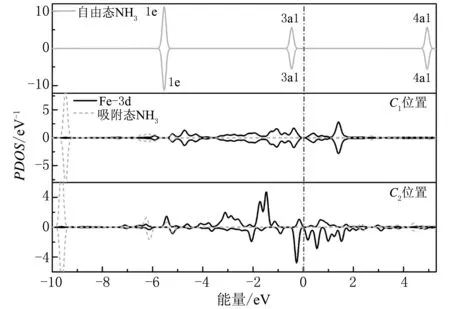
图6 GY-Fe-NH3系统电子分态密度与能量的关系Fig. 6 The relationship between partial density of states and energy of GY-Fe-NH3 system
根据图6分析可知:由于Fe原子与NH3分子的相互作用,C1和C2位置的吸附态NH3分子3a1轨道和1e轨道的电子分态密度峰均会向深能级移动,N原子2p轨道电子态与Fe原子3d轨道电子态发生了杂化交叠;另外,与自由态NH3分子的3a1轨道电子分态密度峰相比,吸附态NH3分子在该轨道的电子分态密度峰存在一定程度的弱化,这种电子结构的变化是由于吸附态NH3分子与GY-Fe表面相互作用产生了电荷转移。通过Bader电荷分析可知:对于掺杂C1和C2位置来说,吸附态NH3分子在GY-Fe表面分别失去了0.141 e 和0.280 e的能量;Fe原子与NH3分子相互作用引起的电荷转移,又会导致界面处电荷的重新排布。从图7所示的GY-Fe-NH3的表面差分电荷密度可以看出,电荷主要积聚在Fe-N键周围。这意味着,NH3分子在GY-Fe表面是以典型化学吸附形式存在的。

(a) C1位置俯视图 (b) C2位置俯视图

(c) C1位置侧视图 (d) C2位置侧视图 图7 GY-Fe-NH3的表面差分电荷密度Fig. 7 Charge density difference of GY-Fe-NH3 surface
4 结论
本文通过第一性原理研究GY和Fe原子掺杂GY表面对NH3分子的吸附特性,得出了以下结论:
(1) Fe原子替代表面C原子能够形成稳定的掺杂系统,Fe原子的掺杂可以有效打破GY表面的化学惰性,提高其表面的化学活性;
(2) 掺杂表面与气体分子NH3之间的电荷转移,以及轨道电子态的杂化交叠结果说明,NH3分子能够以化学吸附方式被束缚在掺杂表面;
(3) Fe原子掺杂单层GY,可用作探测和控制气体分子NH3。
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中学生数理化·中考版(2021年10期)2021-11-22
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
中学课程辅导·教学研究(2017年11期)2017-09-23
新高考·高一物理(2015年6期)2015-09-28
新高考·高一物理(2015年6期)2015-09-28
少儿科学周刊·少年版(2015年1期)2015-07-07
少儿科学周刊·儿童版(2015年1期)2015-07-07
