
Colony stimulating factor 1:friend or foe of neurons?
2022-08-27 07:39LornaBoXuenongBo
中国神经再生研究(英文版) 2022年4期
Lorna Bo,Xuenong Bo
Colony stimulating factor 1 receptor(CSF1R) is a tyrosine kinase receptor primarily expressed on microglia and a small subpopulation of neurons in the central nervous system (CNS),which directly controls the homeostasis,activation,and proliferation of microglia.Its ligands include CSF1 and interleukin-34 (IL-34),which bind to the same region of CSF1R.The two ligands have overlapping functions,however,they also have some differences in signal transduction and induce different transcription profiles.CSF1 and IL-34 are generally expressed by neurons in the CNS,but CSF1 is also expressed by astrocytes.The colony stimulating factors were first characterized by their ability to trigger the differentiation of bone marrow precursor cells into mature myeloid cells but were later found to also act on mature myeloid cells including microglia.In the homeostatic brain,a baseline level of CSF1 helps to maintain microglial roles of synaptic pruning,release of neurotrophic factors,and promotion of brain connectivity.However,over the past decade or so,chronic activation of microglia has been implicated in exacerbating neurodegenerative disorders,including Alzheimer’s disease(AD),Parkinson’s disease,multiple sclerosis(MS),and amyotrophic lateral sclerosis (ALS)(Xu et al.,2021).Yet there have also been studies in contradiction,which showed that in other circumstances,activated microglia were therapeutic and might mitigate neurodegeneration.So,is CSF1 the friend or foe of neurons?
It has been proposed that activation of microglia via CSF1R and other microglial receptors such as TLRs,CX3CR1,and purinoceptors induces different phenotypes depending on the types and stages of the lesion,and the age of the brain,as all these factors into the cytokine milieu.Generally,a proinflammatory M1 phenotype is induced by the stimulation of CSF1R and other receptors in the presence of,for example,pro-inflammatory cytokines,β-amyloid,lipopolysaccharides or myelin debris,and contributes to neurodegeneration.An anti-inflammatory M2 phenotype is produced by the stimulation of CSF1R and other receptors in the presence of antiinflammatory cytokines and contributes to neuroprotection via phagocytosis,proliferation,and remyelination (Figure 1).Upon acute injury,microglia polarize to an M1 phenotype to combat the insult/infection,and subsequently switch to a more M2-like phenotype to enable tissue repair.However,during chronic neurodegeneration,more microglia of M1 phenotype are maintained and,in this way,the microglia mainly exacerbate the disease.
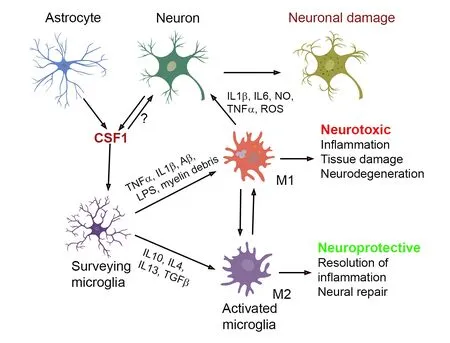
Figure 1|Effects of increased CSF1 release under pathological conditions of the CNS.
We examined levels of CSF1 in the mouse spinal cords of acute and chronic experimental autoimmune encephalomyelitis(EAE) and found that CSF1 was significantly upregulated among neurons,especially the motor neurons,and astrocytes (Gushchina et al.,2018).This corresponded to increased localized proliferation and activation of microglia,which enwrapped the neurons with high-level CSF1.In the chronic EAE mice,the number of large motoneurons expressing CSF1 was significantly lower,perhaps indicating that those motoneurons expressing a high-level of CSF1 had died during the progression of EAE to the chronic stage.Our studies contribute to the growing body of evidence that the CSF1-CSF1R signaling pathway is pivotal to the pathology of EAE,and a potential target for treatments to halt neurodegeneration.Several studies have shown that administration of various CSF1R inhibitors reduced disease severity and slowed its progression,which was associated with a reduction in the number of microglia and infiltrated macrophages,inhibition of myelin-specific T cell responses,and reduced levels of inflammatory factors.A recent study also showed a significant increase in CSF1 and CSF1R levels in the brains of progressive MS patients (Hagan et al.,2020).In the same study administration of a novel CSF1R inhibitor attenuated a disease-associated microglial phenotype and blocked the axonal damage and neurological impairments in an EAE model.
Indeed,many studies in different disease models,such as AD,Parkinson’s disease,ALS,prion disease,and spinocerebellar ataxia type 1,have been performed which show that microglial elimination via CSF1R inhibition ameliorates neurodegeneration and functional recovery (Gomez-Nicola et al.,2013;Martinez-Muriana et al.,2016).Administration of CSF1 to a mouse model of ALS was found to accelerate disease progression,with increased numbers of microglia and upregulated inflammatory cytokines (Gowing et al.,2009).
Yet there is also a body of evidence that implies the opposite,emphasizing the neuroprotective role of CSF1/CSF1R.Jin et al.found that a significant reduction in microglia number (~90%) using the CSF1R inhibitor PLX3397 exacerbated brain infarction and dramatically increased the production of inflammatory mediators by astrocytes–they thus proposed a neuroprotective role of microglia in inhibiting a post-ischemia astrocyte response (Jin et al.,2017).A recent study on a rat hypoxicischemic encephalopathy model showed the intranasal administration of recombinant human CSF1 1 and 24 hours after ischemia reduced the infarcted areas and improved neurobehavioral deficits (Hu et al.,2020).The anti-inflammatory effects of CSF1 are thought to be partly mediated through the CSF1R/PLCG2/PKCε/CREB signaling pathway,presumably,in microglia.Luo et al.(2013) found that CSF1 reduced kainic acid (an excitotoxin)-induced cell loss in the hippocampus when administered systemically before or up to 6 hours after injury.However,they proposed that this occurred via a direct effect on neurons as both CSF1 and IL-34 activated cAMPresponsive element-binding protein signaling in neurons,which enables their survival.Rice et al.(2015) highlighted this microglial dualism of neurotoxicity/neuroprotection by examining the effects of CSF1R inhibition on recovery from diphtheria toxin-induced hippocampal lesions.They found that inhibition of CSF1R post-lesioning improved neuron survival and functional recovery,while inhibition pre-lesioning increased neuron loss and impaired functional recovery.
The studies described above are acute injury models.The neuroprotective effects of CSF1-CSF1R signaling pathways have also been reported in a few EAE and AD models.A recent study showed that administration of CSF1 and IL-34 to the cisterna magna induced the expansion of CD11c+microglia,a population believed to be critical for primary myelination,and resulted in the amelioration of EAE symptoms and decreased demyelination (Wlodarczyk et al.,2018).In an MS model with cuprizoneinduced demyelination,intraperitoneal administration of CSF1 was shown to reduce myelin loss (Laflamme et al.,2018).Such results are contradictory to those reports on the detrimental effects of elevated CSF1 expression in EAE models.Different types of MS models may contribute to the discrepancy of the results as microglia may respond differently to the different methods used to induce demyelination.Furthermore,different treatments,i.e.,administration of CSF1 or IL-34vs.CSF1R inhibitors,may affect different subtypes of microglia such as the CD11c+subtype,which may partly explain the contradictory results observed.
A fewin vitroandin vivostudies support the neuroprotective role of CSF1 in AD models,based on the observation that CSF1-induced activation of microglia enhances their ability to phagocytose β-amyloid and ameliorated memory deficits although Luo et al.(2013)found that the beneficial effects of CSF1 on cognitive function in their hAPP mice are likely independent of Aβ accumulation.Since several other studies demonstrated the beneficial effects of CSF1R inhibition in AD models,it is postulated that the neuroprotective effect of CSF1 is limited to the early stage of the disease.
While the studies mentioned above have largely focused on the effect of CSF1 on microglia,there are conflicting reports as to the expression of CSF1R in neurons.Initial studies by Wang et al.(1999) and another lab using multiple CSF1R antibodies from three companies found CSF1R expression in several neuronal subpopulations.However,these data were somewhat invalidated by Luo et al.(2013) who found that none of the CSF1R antibodies tested was specific as they stained the CSF1R-deficient mouse brains.Luo et al.instead used reporter mouse models,as well asin situhybridization,to detectCsf1rmRNA in neurons.They reproduced some of the findings of Wang et al.(1999)finding that under homeostatic conditions CSF1R was expressed by roughly 1–2% of neurons,mostly located in the hippocampus.Excitotoxicity induced an increase in CSF1R expression,which may implicate a direct role for CSF1 on neurons,rather than via microglial activation.One might speculate that this could have played a role in other studies,such as on the effect of CSF1/CSF1R inhibition in hippocampal lesions (Rice et al.,2015).Overall,very little is known about what direct effects CSF1 might have on neurons.
The role of IL-34 is similarly understudied.Our results showed that IL-34 level did not significantly change in EAE (Gushchina et al.,2018),and this aligns with the observations of Martinez-Muriana et al.(2016) who demonstrated that CSF1 mRNA and protein increased in ALS spinal cords,but IL-34 did not.However,in a prion disease model,CSF1,IL-34 and CSF1R mRNA all increased in the hippocampus and thalamus (Gomez-Nicola et al.,2013).It is possible that IL-34 plays a role in also stimulating CSF1R under certain circumstances,and in some specific regions in the CNS.
In summary,despite the ongoing conflict between studies of the neurodegenerative and neuroprotective roles of CSF1/CSF1R,most research suggests that blocking the CSF1/CSF1R system is neuroprotective in many neurodegenerative models.The supposed mechanism is via the elimination of chronically activated proinflammatory microglia.However,attention must be paid to conflicting evidence concerning the neuroprotective role of CSF1/CSF1R,particularly in AD models and acute brain insults.Future research using different disease models should focus on varying CSF1/CSF1R inhibition by brain region,stages of disease progression,age of the brain,cytokine exposure,etc.This will help elucidate the spatiotemporal divergence of microglial phenotypes and functions.Further research should also aim to investigate the role,if any,of IL-34 and the potential direct effects of CSF1 on neurons.Such knowledge will enable the development of clinical studies targeted at the CSF1/CSF1R pathway.
We thank Miss Longyan Sun for the artwork in Figure 1.We apologize to the researchers for not being able to cite their research described in the article due to the limit of space.
The present work was supported by Foresight Inc.(to XB).No conflicts of interest exist between Foresight Inc.and publication of this work.

- 中国神经再生研究(英文版)的其它文章
- Towards a comprehensive understanding of p75 neurotrophin receptor functions and interactions in the brain
- Microglia regulation of synaptic plasticity and learning and memory
- Stroke recovery enhancing therapies:lessons from recent clinical trials
- Functional and immunological peculiarities of peripheral nerve allografts
- MicroRNA expression in animal models of amyotrophic lateral sclerosis and potential therapeutic approaches
- Significance of mitochondrial activity in neurogenesis and neurodegenerative diseases