
Microglia regulation of synaptic plasticity and learning and memory
2022-08-27 07:39JessicaCornellShelbiSalinasHouYuanHuangMiouZhou
中国神经再生研究(英文版) 2022年4期
Jessica Cornell ,Shelbi Salinas ,Hou-Yuan Huang ,Miou Zhou
Abstract Microglia are the resident macrophages of the central nervous system.Microglia possess varied morphologies and functions.Under normal physiological conditions,microglia mainly exist in a resting state and constantly monitor their microenvironment and survey neuronal and synaptic activity.Through the C1q,C3 and CR3“Eat Me”and CD47 and SIRPα“Don’t Eat Me”complement pathways,as well as other pathways such as CX3CR1 signaling,resting microglia regulate synaptic pruning,a process crucial for the promotion of synapse formation and the regulation of neuronal activity and synaptic plasticity.By mediating synaptic pruning,resting microglia play an important role in the regulation of experience-dependent plasticity in the barrel cortex and visual cortex after whisker removal or monocular deprivation,and also in the regulation of learning and memory,including the modulation of memory strength,forgetfulness,and memory quality.As a response to brain injury,infection or neuroinflammation,microglia become activated and increase in number.Activated microglia change to an amoeboid shape,migrate to sites of inflammation and secrete proteins such as cytokines,chemokines and reactive oxygen species.These molecules released by microglia can lead to synaptic plasticity and learning and memory deficits associated with aging,Alzheimer’s disease,traumatic brain injury,HIV-associated neurocognitive disorder,and other neurological or mental disorders such as autism,depression and post-traumatic stress disorder.With a focus mainly on recently published literature,here we reviewed the studies investigating the role of resting microglia in synaptic plasticity and learning and memory,as well as how activated microglia modulate disease-related plasticity and learning and memory deficits.By summarizing the function of microglia in these processes,we aim to provide an overview of microglia regulation of synaptic plasticity and learning and memory,and to discuss the possibility of microglia manipulation as a therapeutic to ameliorate cognitive deficits associated with aging,Alzheimer’s disease,traumatic brain injury,HIV-associated neurocognitive disorder,and mental disorders.
Key Words:aging;Alzheimer’s disease;cognitive deficits;experience-dependent plasticity;learning and memory;mental disorders;microglia;synaptic plasticity;synaptic pruning
Introduction
As the resident immune cells in the central nervous system(CNS),microglia play an important role in the regulation of neuronal activity and synaptic plasticity.Microglia are the immune and neuronal support cells of the brain,surveying and“sensing”the microenvironment via microglial processes(Reemst et al.,2016;Elmore et al.,2018).Synaptic pruning is necessary for precise neuronal circuitry during development as well as synaptic plasticity in the adult brain via the C1q,C3 and CR3 complement signaling or the CD47 and signal-regulatory protein α (SIRPα) signaling pathways (Miyamoto et al.,2016;Sellgren et al.,2017;Lehrman et al.,2018;Elberg et al.,2019).Resting microglia have important physiological functions in the regulation of neuronal activity,synaptic transmission,and the formation,modification or elimination of synaptic structures (Akiyoshi et al.,2018).Microglia frequently interact with neurons.Through signaling molecules such as brainderived neurotrophic factor (BDNF) and transforming growth factor-β and complement proteins including C1q and CR3,microglia are able to communicate with neurons to mediate neuronal function (Hong et al.,2016;Wang et al.,2020).This microglia-neuron communication is not only critical in brain development and maturation,but also for synaptic plasticity and learning and memory in the mature brain (Figure 1).

Figure 1|Resting microglia regulate synaptic pruning,synaptic plasticity and cognition in the healthy brain.
Microglial activation or dysfunction is associated with the progression of cognitive deficits in normal aging,neurodegenerative and neurological diseases such as Alzheimer’s disease (AD),traumatic brain injury (TBI) and HIV-associated neurocognitive disorder (HAND),and mental disorders including autism,stress and depression (Elmore et al.,2018;Krukowski et al.,2018;Lee et al.,2018;Zuckerman et al.,2018;Li et al.,2021).For example,microglia in the aging brain can become“primed”where they are constantly activated at low levels and do not fully reenter the resting state(Koss et al.,2019).This results in an increase in the number of microglia,proinflammatory cytokines and phagocytic activity which result in decreased synaptic plasticity and compromised learning and memory (Tichauer et al.,2014;Elmore et al.,2018;Zöller et al.,2018).AD studies show decreased microglial phagocytic activity in direct relation to deficient triggering receptor expressed on myeloid cells 2 (TREM2) and interleukin 33 (IL-33).The lack of microglia phagocytic activity results in the accumulation of plaques and the dystrophy of neurons (Fu et al.,2016;Lee et al.,2018).Dysfunctional microglia also lead to enhanced proinflammatory cytokines and inflammation and the destruction of neuronal function in TBI,HAND and mental disorders,which cause synaptic plasticity and memory deficits (Makinde et al.,2017;Krukowski et al.,2018;Rawat et al.,2019;Worthen et al.,2020;Li et al.,2021).Although microglia have been studied in multiple brain pathologies,their contribution to learning and memory deficits,the interaction with neurons in these diseases,and the molecular and cellular mechanisms involved still require further investigation.By summarizing the recent studies about how microglia regulate synaptic plasticity and cognitive function under normal and diseased conditions,we hope this review can strengthen the understanding of the role of microglia in normal learning and memory and experiencedependent plasticity,and potentially aid in the treatment of cognitive deficits associated with different neurological or mental disorders by targeting microglia or microglia-related signaling pathways.
Search Strategy and Selection Criteria
Studies cited in this review were published from 2010 to 2021 (except one reference from 1998),with a predominant emphasis from 2016 to 2021.All studies cited were searched on PubMed database using the following keywords:microglia,synaptic plasticity,synaptic pruning,neuronal activity,learning and memory,development,adult.All studies were cited due to their relevance to the review.
Microglia Regulation of Synaptic Pruning
Microglia play an important role in the identification and removal of unnecessary neural connections.Synaptic pruning is the process of synapse removal that occurs during postnatal development and throughout adulthood (Brioschi et al.,2020;Liu et al.,2021).This process promotes the refinement of neural circuits and increases neuronal network efficiency(Liu et al.,2021).The signals microglia and neurons utilize to regulate synaptic pruning are not yet entirely known.However,it is important to determine the particular indications or instructive signals that recruit microglia to certain synapses for pruning,so microglia can identify and remove unnecessary neural connections via these specific signaling pathways.
In the healthy brain,the classical complement cascade functions as a sort of“tagging”mechanism for microglia pruning (Table 1) (Györffy et al.,2018;Datta et al.,2020).Complement proteins specifically localize and bind to apoptotic,immature or weak developing synapses in the CNS.These synapses are then recognized by complement receptors and consequently engulfed.C1q,the initiating protein of the cascade,and another complement protein,C3,are predominantly produced by microglia or astrocytes,and localize to the appropriate synapses (Fonseca et al.,2017;Wu et al.,2019).C1q and C3 are recognized by the complement receptor CR3 which is exclusively expressed on microglia,mediating the engulfment of synapses (Hong et al.,2016;Anderson et al.,2019).C1q presynaptic tagging is correlated with markers of apoptosis,such as cleaved caspase-3 or annexin V.The number of apoptotic markers in the synapse equates to the localization of C1q,which prompts synaptic pruning (Györffy et al.,2018).Because of the important role of complement system in synaptic pruning,C1q,C3 or CR3 knockout (KO) mice show defects in pruning and synaptic connectivity (Shi et al.,2015;Hong et al.,2016;Anderson et al.,2019;Wang et al.,2020).

Table 1|Signaling involved in microglia-mediated synaptic pruning
A recent study by Wang et al.(2020) shows that the C1qdependent complement pathway is strongly involved in synapse elimination by microglia,and CD55,an inhibitor of complement pathways,can disrupt this microglia and C1q-dependent complement pathway in engram cells which store memory.Because microglia are inhibited from eliminating synapses between engram cells in the dentate gyrus of healthy adult mice,reactivation of engram cells is increased in CD55-treated mice (Wang et al.,2020).It is worth noting that synapse elimination is an activity-dependent process,and the complement-mediated“tagging”allows for elimination specificity by microglia.As a result,active synapses are selectively maintained while nonessential synapses are eliminated (Györffy et al.,2018).For example,a typical postsynaptic dendrite is initially innervated by several axons.Through pruning,the inputs that are relatively weak are removed while the more active inputs are strengthened and maintained (Weinhard et al.,2018).In contrast to theimportant role of complement in synaptic pruning,Cr3KO mice does not affect the partial phagocytosis of presynaptic structures such as axons and synaptic boutons by microglia,a process known as trogocytosis (Weinhard et al.,2018).This suggests that classical complement signaling may not be necessary for trogocytosis.
Another“Eat Me”signal that promotes synaptic pruning is phosphatidylserine (PS).PS exposure typically occurs on apoptotic or injured dendrites (Scott-Hewitt et al.,2020).TREM2 is a cell-surface receptor on microglia that promotes synaptic pruning.PS can be recognized by TREM2 and consequently be targeted for elimination (Filipello et al.,2018;Sapar et al.,2018;Shacham-Silverberg et al.,2018).Bothin vivoandin vitrotesting show that PS and TREM2 interaction can regulate synaptic pruning in the hippocampus and dorsal lateral geniculate nucleus (Scott-Hewitt et al.,2020).Furthermore,Trem2KO mice have a decrease in microglial density,an increase in neuronal spine density,and impaired connectivity in the hippocampus (Filipello et al.,2018).
Microglia also regulate phagocytosis and synapse elimination through the fractalkine receptor C-X3-C motif chemokine receptor 1 (CX3CR1).CX3CR1 is highly expressed in microglia,and is considered to be essential to microglia and neuron communication (Bolós et al.,2018).Its ligand,CX3CL1,is expressed by neurons (Zhang et al.,2018).The CX3CR1/CX3CL1 signaling contributes to synaptic pruning,allowing microglia to know which synapses need to be eliminated.The interaction between CX3CR1 and CX3CL1 also regulates the engulfment of oligodendrocyte progenitor cells (OPCs) by microglia during development.CX3CR1KO/KO:GFP mice show a reduction in engulfment of OPCs compared to CX3CR1:GFP wild-type (WT) mice although there is no change in microglia number or frequency of OPC contact (Nemes-Baran et al.,2020).Another study shows that synapse engulfment is impaired in microglia lacking CX3CR1 or deficiency of the ligand CX3CL1,and whisker cauterization and trimming results in reduced synaptic activity in the barrel cortex (Gunner et al.,2019).
In contrast to C1q,C3 and CR3 mediated microglial pruning,another class of molecules,referred to as“Don’t Eat Me”signals,act as“stop”signals and protect active synapses from being engulfed.Microglia express receptors that recognize these“Don’t Eat Me”signals.CD47 is a particular“Don’t Eat Me”protein.It binds to its receptor SIRPα which is highly expressed on microglia during peak pruning periods in development,and inhibits the phagocytosis of synapses(Sato-Hashimoto et al.,2019).In apoptotic or damaged cells,the“Don’t Eat Me”signals are downregulated,and as a result,complement proteins enable the engulfment of the damaged cells (Lehrman et al.,2018).Because the“Don’t Eat Me”signals inhibit synaptic pruning,the loss of CD47 or SIRPα results in excessive pruning during postnatal development (Lehrman et al.,2018).CD47 is localized to the more active synapses,suggesting that synaptic pruning is activity dependent and CD47 potentially protects highly active synapse populations from targeting by microglia-mediated synaptic pruning.Apart from microglia,SIRPα-CD47 binding also leads to increased phagocytosis in macrophages (Elberg et al.,2019).Another interaction that may be considered a“Don’t Eat Me”signal is the CD200/CD200R pathway.CD200 is expressed by neurons and CD200R is expressed by microglia (Sun et al.,2020).Cd200KO mice display an increase in phagocytosis compared to WT mice in the presence of amyloid beta (Lyons et al.,2017).Via these“Don’t Eat Me”signals,it can prevent the engulfment of strong and active synapses that contribute to learning and memory.
Overall,these above studies establish that synaptic pruning by microglia is a highly regulated process.Synaptic pruning begins in development and occurs throughout adulthood in multiple brain regions,and it is vital that the appropriate synapses are removed while others are strengthened and maintained through microglia regulated synaptic pruning.
Microglia,Neuronal Activity and Synaptic Plasticity
In physiological conditions,motile microglial processes are constantly monitoring their microenvironment.Through neuron and microglia interactions,microglia are able to regulate neuronal activity,synapse formation and survival,and synaptic circuit remodeling (Badimon et al.,2020;Saw et al.,2020;Sun et al.,2020).Synaptic plasticity is an ability of the CNS to modify synapses and neural connections in response to synaptic activity and sensory and motor experiences.Microglia are critically involved in the modulation of synaptic plasticity,including long-term potentiation (LTP) and longterm depression (LTD) (Table 2) (Raghuraman et al.,2019;Kim et al.,2020).
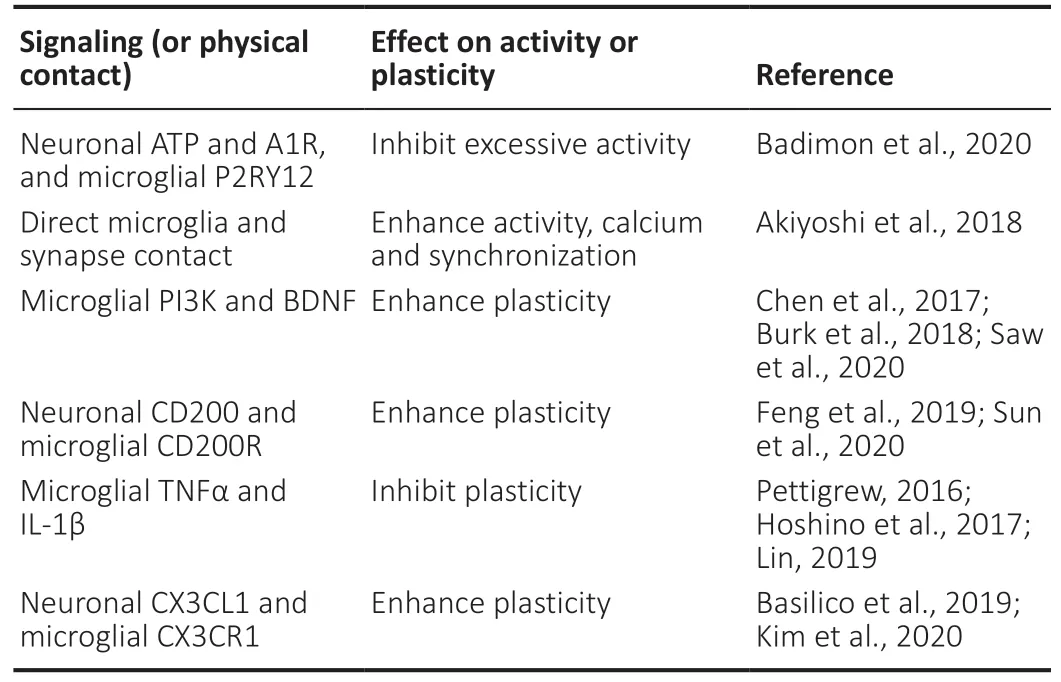
Table 2|Microglia regulation of neuronal activity and synaptic plasticity
Microglia and neuronal activity
Microglial processes are constantly moving and surveying their environment.Through their surveillance,microglia detect which synapses are strong,weak or inactive,and determine which synapses to remove while others are conserved and strengthened.During this process,neuronal activity contributes to the ability of microglia to regulate synaptic pruning and,ultimately,synaptic plasticity.For example,in the visual system,reducing neuronal activity after light deprivation increases the number of microglia at dendritic spines that eventually diminish in size,indicating that microglia-mediated pruning eliminates synapses that are less active (Tremblay et al.,2010;Pan and Monje,2020).
Microglia motile processes physically contact neuronal synapses.As microglia survey and“sense”their microenvironment,they are prepared to respond to any changes or disruptions to homeostasis.For example,neurons exhibit a local release of adenosine triphosphate (ATP) upon synaptic activation (Lalo et al.,2016).ATP release functions as a chemoattractant for microglial processes via purinergic receptors.Resting microglia highly express the purinergic receptor,P2RY12,which controls ATP/ADP-dependent chemotaxis and motility of microglia,and promotes neuron and microglia communication (Sipe et al.,2016;Peng et al.,2019;Badimon et al.,2020).ATP concentration must reach a threshold for P2RY12 to recognize ATP and recruit microglia to the synapse,and blocking P2RY12 inhibits the ability of microglia to sense ATP and ADP (Badimon et al.,2020).Microglia break down ATP using the enzymes CD39 and CD73 to produce adenosine (ADO),which then acts on the adenosine receptor,A1R,located on active neurons to prevent neuronal hyperactivation.Through the neuron-microglia interaction,microglia are able to prevent excessive neuronal activation via ATP-AMP-ADO-A1R-dependent feedback(Badimon et al.,2020).Microglia-deficient mice exhibit exaggerated behavioral responses,such as seizures,upon neurostimulation with kainic acid,picrotoxin,or dopamine D1 receptor agonist (Badimon et al.,2020).Through microgliamediated suppression,microglia can reduce seizure severity by detecting excessive neuron excitability and decreasing neuronal firing rates (Badimon et al.,2020).These findings suggest that microglia are critical in the regulation of neuronal activity,specifically in preventing neuron hyperactivation via a negative feedback mechanism.It serves as a protective mechanism in that excessive hyperactivity is damaging to neurons and can result in neuronal death and synaptic loss,which leads to reduced plasticity (Lepeta et al.,2016;Badimon et al.,2020).The microglia-mediated negative feedback mechanism contributes to homeostasis of neuronal activity and maintains synaptic integrity and plasticity by modulating neuronal activity.
The frequent contact between microglia and synapses also has a functional significance and can promote local network synchronization followed by an increase in neuronal activity(Akiyoshi et al.,2018).In vivotwo photon Ca2+imaging with the use of the Iba1-EGFP transgenic mice demonstrates that neuronal synapse activity is increased when they contact microglia (Akiyoshi et al.,2018).When the normal function of microglia is abolished by microglia activation with lipopolysaccharide or by ablation of microglia in Iba1-tTA/tetO-DTA double transgenic mice,the synchronization of L2/3 neurons located in close proximity to one another is decreased,indicating that microglia can regulate neuronal activity and synchronization.This study suggests that interactions between resting microglia and synapses in the healthy brain helps to synchronize local populations of neurons,which provides a plausible explanation on how alterations in immune status may change synaptic plasticity(Akiyoshi et al.,2018).
Microglia and synaptic plasticity
Resting microglia promote long-term plasticity and synaptic tagging and capture (STC) under normal conditions(Raghuraman et al.,2019).The brain has constant changes in the structure and remodeling of neural circuits.This contributes to synaptic plasticity such as the stability of LTP,which is believed to be the cellular mechanism of learning and memory (Poo et al.,2016;Lisman et al.,2018).The STC hypothesis is first demonstrated by Frey &Morris who show that early phase of LTP from weak stimulation can possibly become late phase LTP if strong stimulation from another pathway is applied to the same population of neurons within a specific period (Frey and Morris,1998).The beginning weak stimulation results in the formation of short-lasting“synaptic tags”to some synapses.Strong stimulation from the other pathway then activates the dopaminergic inputs and further leads to increase in plasticity related proteins in these synapses (Okuda et al.,2020).The STC provides the basic framework for the transformation of short-term plasticity into long-term plasticity (Nomoto and Inokuchi,2018),and importantly,microglia have a specific role in STC.Clodronate treatment specifically eliminates microglia without affecting the viability of other cell types (Wang et al.,2018).Clodronate treatment during the induction or early phase of LTP prevents the late LTP expression,suggesting a critical role of microglia in early STC and LTP induction.Disrupting microglia with clodronate after the establishment of late phase LTP has no effect on STC,as it does not block late LTP expression(Raghuraman et al.,2019).These results demonstrate that microglia play a significant role in setting the synaptic tags during the early phase of activity dependent plasticity which promote the stability of LTP.Once the stabilized LTP is established,microglia are no longer required to maintain the plasticity and STC.
In addition to LTP,LTD,which involves the reduction of synaptic transmission,potentially depends on microglial fractalkine signaling.CX3CR1/CX3CL1 signaling contributes to changes in microglia morphology and expression of immune proteins such as chemokines and chemokine receptors (Gyoneva et al.,2019).A decrease in fractalkine gene expression and protein concentration in R6/1 transgenic mice,a mouse model of Huntington’s disease,results in striatal synaptic impairment.LTD induction by theta-burst stimulation in R6/1 transgenic mice decreases the amplitude of striatal field postsynaptic current and leads to synaptic plasticity deficits(Kim et al.,2020).In WT mice treated with minocycline,an inhibitor of microglial activation,LTD is not induced.However,administration of fractalkine is able to restore LTD (Kim et al.,2020).Furthermore,deficient microglia and neuron signaling in the CA1 of the hippocampus during brain development is associated with irregular pre-synaptic maturation.In particular,Cx3cr1KO mice have reduced glutamate release and excitatory postsynaptic currents (Basilico et al.,2019).This demonstrates the importance of microglia and neuron communication via CX3CR1/CX3CL1 for proper synaptic function and maturation.
In the healthy brain,there is a particular balance between proinflammatory and anti-inflammatory markers released by microglia that,if disrupted,can affect synaptic plasticity (Golia et al.,2019).In particular,treatment with lipopolysaccharide increases inflammatory marker expression in microglia,such as tumor necrosis factor α (TNFα) and IL-1β (Lin et al.,2019),and decreases BDNF levels and phosphorylation of the AMPA(α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid)receptor subunit GluR1 (Golia et al.,2019).Overexpression of TNFα results in an increase in LTP for a brief period after induction via electrical stimuli (Pettigrew et al.,2016),suggesting that raised levels of TNF-α may cause synaptic networks to be hyperexcitable.However,spatial memory deficits in the Morris Water Maze are present in TNFα Tg rats although there are no differences in the loss of cortical and hippocampal neurons (Pettigrew et al.,2016).Compared to TNF-α,another microglia released proinflammatory protein,IL-1β,impairs LTP in CA1.Upon electrical stimulation application in the stratum radiatum of the CA1 and CA3 regions of the hippocampus,NMDA receptor-dependent LTP at Schaffer collateral-CA1 synapses is impaired with IL-1β treatment (Hoshino et al.,2017).These results suggest that the microglia released proinflammatory proteins exert variable effects on LTP.The exact effect of microglia in LTP strengthening or impairment depends on the molecule released by microglia.
The CD200 and CD200R signaling pathway can contribute to synaptic plasticity and help to keep microglia in a resting state by inhibiting the release of proinflammatory mediators (Jiang et al.,2016;Xie et al.,2017;Oria et al.,2018;Feng et al.,2019).Overexpression of neuronal CD200 in the hippocampus inhibits microglial activation,improves dendritic density,and enhances synaptic plasticity and cognitive function (Feng et al.,2019).Injection of CD200Fc,a CD200R agonist,activates the CD200/CD200R signaling pathway and increases dendritic spine density and functional recovery after stroke-induced brain damage (Sun et al.,2020).These results suggest that the CD200/CD200R pathway regulates synaptic plasticity through the inhibition of microglia-induced proinflammatory response.Microglia can also modulate synaptic transmission by BDNF expression via PI3K/BDNF signaling,which contribute to enhanced synaptic plasticity.BDNF,which has been shown to increase dendritic spine plasticity in the adult cortex (Chen et al.,2017;Burk et al.,2018;Rauti et al.,2020),is a downstream effector of microglial phosphatidylinositol 3-kinase (PI3K) (Saw et al.,2020).Microglial PI3K is involved in synaptic plasticity through the modulation of BDNF expression in microglia.Upregulation of PI3K can lead to an upregulation of BDNF andvice versa(Saw et al.,2020).Upon phosphorylation,AKT stimulates the phosphorylation of CREB,an important signaling in LTP and learning and memory (Sehgal et al.,2018)which regulates the expression of BDNF gene expression in microglia.Mature BDNF then binds to tropomyosin sensitive receptor kinase B receptors located on neurons,which trigger signaling cascades such as PI3K,mitogen-activated protein kinase,or phospholipase C in neurons.Each of these signaling cascades has been shown to be linked to synaptic plasticity(Jung et al.,2017;Li et al.,2020a;Sun et al.,2020).As a result,microglia PI3K signaling influences the expression and secretion of BDNF,which then promotes axonal branching,dendritic growth,and synapse refinement in an activitydependent manner,and therefore enhances synaptic plasticity.Altogether,microglia exhibit variable effects on synaptic plasticity based on the dynamics between proinflammatory and anti-inflammatory cytokines released.The microglial proinflammatory or anti-inflammatory molecules,including PI3K,BDNF,CREB,CD200,TNFα and IL-1β,contribute to the enhancement or inhibition of synaptic plasticity.Resting microglia promote long-term plasticity and play a critical role in early STC and LTP induction,which are mediated by the microglial CREB and PI3K/BDNF signaling.Proinflammatory cytokines released from microglia have variable effects on LTP and plasticity.The CD200 pathway inhibits microgliainduced inflammatory response and enhances synaptic plasticity.Overexpression of TNFα results in an increase in LTP for a brief period while IL-1β impairs LTP in CA1.Together,these results demonstrate the diverse effects of microglia on synaptic plasticity are based on its proinflammatory or antiinflammatory profile as well as the state of microglia.
Role of Microglia in Learning and Memory and Experience-Dependent Plasticity
In the adult brain,synapse remodeling is a continued process that changes synaptic connectivity and is essential for memory encoding in the neural circuit.Mechanisms that contribute to synapse remodeling is critical to the flexibility of learning and memory.As mentioned above,microglia make activitydependent contact with the synapses to regulate synaptic density and connectivity (Madry et al.,2018),and several recent studies have shown that microglia not only regulate neural ensemble connectivity and memory strength,but also memory quality (Nguyen et al.,2020;Wang et al.,2020).Besides learning and memory,microglia are also required for some forms of cortical plasticity in an experience-dependent manner (Table 3) (Miyamoto et al.,2016;Sipe et al.,2016).

Table 3|Microglia regulation of learning and memory and experience-dependent plasticity
Microglia and learning and memory
The involvement of microglia in learning and memory has been reported in multiple studies,with majority of these studies reporting a negative role of microglia in learning and memory under inflammation and pathological conditions(Feng et al.,2019;Li et al.,2020b;Mohammadi et al.,2020;Worthen et al.,2020;Xie et al.,2020;Zhang et al.,2020a;Liao et al.,2021).Here we review three recent studies showing the specific role of microglia in memory quality (i.e.the ability to discriminate in similar contexts) and the strength of remote memory.Microglia are involved in the control of memory quality via IL-33 signaling (Nguyen et al.,2020).Loss of IL-33 receptors on microglia or neuronal IL-33 reduces newborn neuron integration and impairs synaptic plasticity and fear memory precision.IL-33 is an interleukin-1 family regulatorfor microglial activation,and in the adult hippocampus,IL-33 is mainly expressed in neurons in an experience-dependent manner.The IL-33 signaling induces microglia engulfment of the extracellular matrix (ECM),a structure involved in neuronal plasticity and learning and memory (Vainchtein et al.,2018).In this study the transgenic mice IL-33mCherry/+and IL-33 conditioned knockout (cKO) are used to study the IL-33 expression or to remove IL-33 in neurons,and the IL1RL1-cKO mice are used to remove the IL-33 receptor specifically in microglia.Microglia IL1RL1-cKO mice show limited increase in the number of spine head filopodia compared to control mice,suggesting IL-33 signaling from neuron to microglia promotes new spine formation.Mice with IL-33 KO show intact fear memory and no significant differences in anxiety compared to control mice,however,these mice show increased freezing in an unconditioned context 14 or 28 days post fear conditioning training (Nguyen et al.,2020),suggesting that microglia regulate contextual memory quality via the IL-33 signaling.
Microglia also regulate the formation and stability of synapses which further contribute to neurogenesis in learning and memory (Bennett and Molofsky,2019;Wang et al.,2020).Neurogenesis continues to occur in specific brain regions throughout adulthood.The newborn neurons are involved in hippocampus-dependent learning and memory (Diaz-Aparicio et al.,2020).Specifically,neurogenesis regulates forgetting during adulthood and infancy,since hippocampal neurogenesis levels are high during infancy and infant memories are rapidly forgotten (infantile amnesia) while decreasing neurogenesis after memory formation has been shown to mitigate forgetting(Akers et al.,2014;Tran et al.,2019).Microglia mediate forgetting by playing an active role in adult neurogenesis via phagocytosis (Diaz-Aparicio et al.,2020;Wang et al.,2020).In adulthood,most newborn neurons undergo apoptosis after formation,which are identified and removed by microglia(Diaz-Aparicio et al.,2020).Chronic deficiency in microglial phagocytic function reduces adult hippocampal neurogenesis,whereas acute deficiency briefly increases neurogenesis (Diaz-Aparicio et al.,2020).When treated with the pro-neurogenic drug memantine,CX3CR1GFP/+mice show enhanced adult neurogenesis and colocalization of PSD95 with the lysosome marker Lamp1 within microglia,indicating an increase in microglia-mediated phagocytosis which significantly promotes forgetting 35 days after contextual fear conditioning training.In comparison,microglia depletion with PLX treatment prevents memantine-treatment induced forgetting after contextual fear conditioning training,possibly through the increased reactivation rate of engram cells which prevents the forgetting of previously learned memory after microglia depletion (Wang et al.,2020).Microglia-mediated forgetting can also be rescued by expressing CD55 (also known as decayaccelerating factor) which is an inhibitor of complement pathways,suggesting that microglia regulate forgetting of the remote memories mainly via the complement-mediated synaptic elimination.
The repopulation of microglia after its ablation has been shown to improve short-term memory and learning tasks.The morphology and activity of microglia are critical to learning and memory.While acute microglia ablation via the administration of diphtheria (DT) to theCx3cr1-Dtrtransgenic Wistar rat has no effect on short-term memory in both novel object recognition (NOR) and novel place recognition tasks,the newly repopulated microglia with ameboid morphology are associated with memory enhancement and increased numbers in mature neurons in the hilus.Cx3cr1-Dtrrats show increased positive discrimination ratio compared to WT in both NOR and novel place recognition tasks 7 days after DT (De Luca et al.,2020).The results suggest that rats with the repopulated microglia have better short-term memory.Elevated expression of microglia recruitment marker Cxcl10 along with complement initiation protein C1q and C3 are presented in the hippocampus ofCx3cr1-Dtrrats 7 days post DT (De Luca et al.,2020).The repopulation of microglia also significantly enriches the numbers of mature bifurcated spines,which typically is associated with improved spine efficacy (Gipson and Olive,2017;De Luca et al.,2020).Altogether,these results suggest that microglia play a complex role in memory and learning through synaptic pruning and neuronal remodeling.
Microglia and experience-dependent plasticity
Microglia are able to respond to alterations in sensory inputs and play a major role in synaptic remodeling (Miyamoto et al.,2016;Arcuri et al.,2017).Disruption of microglia during the critical period can significantly impact neural development(Miyamoto et al.,2016;Hanamsagar et al.,2018).The barrel cortex is located in the rodent primary somatosensory cortex processing signals from the vibrissae.A recent study shows that microglia are involved in the modulation of neuronal structural remodeling in response to alteration in whisker sensory input (Kalambogias et al.,2020).Microglia undergo morphological changes from a resting state into altered state with retracted processes and expanded soma size during sensory deprivation via whisker deprivation.Sensory restoration from whisker regrowth reverts these altered microglia morphological changes back to age-matched control mice (Kalambogias et al.,2020),indicating that microglia may be recruited to neuronal structural remodeling in response to alteration in sensory input during developmental critical periods.
Besides barrel cortex plasticity,microglia also play an important role in visual experience-dependent plasticity (Wang et al.,2016).Microglia are critical for synaptic transmission and maintaining synaptic structure under normal vision function in adolescent mice.Sustained ablation of microglia leads to progressive degeneration of the photoreceptor synapses and ultimately results in the progressive deterioration of the retinal photon response,indicating the essential role of microglia in maintaining underlying synapses for normal vision.Under monocular deprivation condition,microglia show altered morphology,motility and phagocytosis as well as interactions with synapses.P2Y12 receptor is expressed in microglia and mediates process motility.The disruption of P2Y12 receptor reshapes the response of microglia to monocular deprivation and overturn ocular dominance plasticity,demonstrating the significant role of P2Y12 in microglial modulation of ocular dominance plasticity(Sipe et al.,2016).
Microglial surveillance and regulation of synaptic plasticity in the mouse visual cortex have been studied between wakeful and anesthetized arousal states.Anesthetized mice have increased microglia process arbors compared to awake mice.Elevated response of microglia to focal tissue injury during anesthesia is observed (Stowell et al.,2019),suggesting that wakefulness exhibits a primary inhibitory effect on microglial dynamics.Norepinephrine (NE) is a potent neurotransmitter in the CNS and modulates plasticity,learning,attention to salient stimuli and sensory processing.NE is also a powerful mediator for wakefulness (Xing et al.,2016).Microglial β2-adrenergic receptor (β2-AR) signaling is a critical regulator for microglia surveillance and injury response in the visual cortex of awake mice.When monocular deprivation is used to investigate the role of microglia β2-AR NE signaling in synaptic plasticity,clenbuterol,a selective agonist of β2-AR which chronically activates β2-AR signaling,prevents ocular dominance plasticity.Ablation of β2-AR in microglia abolishes the morphological changes between awake and anesthetized state,demonstrating that microglial surveillance and regulation of synaptic plasticity are modulated by noradrenergic tone fluctuations between wakeful and anesthetized states.Altogether,these results demonstrate the important role of microglia in visual and somatosensory experience-dependent plasticity.
Role of Microglia in Plasticity and Memory Deficits Associated with Different Disorders
While microglia have a positive role in the CNS by maintaining immune homeostasis and playing a crucial role in synaptic plasticity and cognitive function,it can also disrupt neuronal plasticity and cognitive function in different neurological or mental disorders (Table 4andFigure 2).Microglia exist primarily in two states:resting and activated.In the resting state,they project ramifications that sense its environment for changes and secrete neurotrophic factors,which contribute to CNS homeostasis and neuronal plasticity.If changes such as neuronal injury are sensed,microglia will take on the active state,transition into an amoeboid morphology and secrete proinflammatory cytokines and reactive oxidative species(Spencer et al.,2016;Koss et al.,2019).When the response is sufficient,the microglia will transition back into the restingstate.Dysfunction occurs when inflammation becomes constant or chronic,and the microglia do not fully inactivate.Microglia are considered primed at this stage,and primed microglia are easily activated,producing a faster and abnormal hyperactive response to neuronal injury (Koss et al.,2019).

Table 4|Microglia regulation of cognitive deficits

Figure 2|Activated microglia contribute to learning and memory deficits in different disorders.
Microglia and aging
As an individual ages,the microglia transition into the hypersensitive primed phenotype.This chronically activated microglia promote neuronal death and neurodegeneration,which negatively impact synaptic plasticity.Aged mice in comparison to young mice show CA1 LTP impairment in hippocampal slices,as well as spatial memory deficits in the Morris Water Maze (Elmore et al.,2018).Iba1+cell population and Iba1+/CD68+cell expression are significantly increased in the aged mice,indicating an increase in primed microglia in aged mice compared to young mice.There is also a morphological difference between aged and young mice microglia,in that the aged mice show shorter and thicker processes compared to young mice.Eliminating the old microglia from the CNS with a CSF1R inhibitor,PLX3397,and allowing microglia to repopulate reverse the morphology differences in the aged mice to comparative young levels(Elmore et al.,2018).Aged mice show improvement in LTP and spatial memory to that of young control levels after microglial repopulation,suggesting that the primed microglia contribute to the impairment of synaptic plasticity and learning and memory associated with aging (Elmore et al.,2018).
TREM2 receptor is exclusively expressed by microglia in the CNS,and TREM2 signaling promotes microglia phagocytic activation (Filipello et al.,2018).The role of TREM2 in microglial dysfunction varies between aging and AD.Whereas TREM2 deletion in microglia normally has negative effects on certain neurodegenerative disorders like AD,it has been reported that TREM2 deletion improves LTP and learning and memory in normal aging mice,and thus has a beneficial effect for aging.Young WT mice andTrem2–/–mice do not show a significant difference in cognitive tests performance such as open-field test,elevated plus maze,and Morris Water Maze,as well as hippocampal LTP.In contrast,agedTrem2–/–mice have improved cognitive function and hippocampal LTP compared to aged WT mice,indicating that TREM2 expression negatively affects synaptic plasticity in aged mice.AgedTrem2–/–mice also show increased dendritic spines in the CA1 hippocampal region,whereas in typical aged mice,dendritic spines are significantly reduced,indicating a protective effect of TREM2 deletion on dendritic spine loss in the aging brain(Qu and Li,2020).
In addition to the effect of IL-33 on modulating memory generalization in young mice,IL-33 is also involved in the deficits of precision memory in aged mice.Neuronal IL-33 and precision memory are decreased in aged mice.Because neuronal IL-33 drives microglial engulfment of the ECM,decreased IL-33 with aging leads to impaired ECM engulfment and the accumulation of presynaptic ECM,and as a result,the deficits in synaptic plasticity and memory (Nguyen et al.,2020).The IL-33 gain-of-function in aged mice mitigates the age-related decrease in spine density.Therefore,via the neuronal-microglial IL-33 signaling,microglia regulate ECM clearance and spine formation,which are required for the precision of remote memories,and the impaired IL-33 signaling in aged mice leads to impaired memory precision with aging (Nguyen et al.,2020).
Microglia and Alzheimer’s disease
Aging is a large risk factor for the development of AD,and chronic inflammatory responses induced by exaggerated microglia response to Aβ plaques have been associated with the cognitive deficits in AD patients.Similar to the effect of IL-33 on aging,microglia-associated cognitive deficits in AD mice can be improved by IL-33 treatment.APP/PS1 (human amyloid precursor protein/mutant human presenilin 1) mice show LTP impairment in the CA1 region of the hippocampus as well as reduced freezing behavior (Fu et al.,2016).Administration of IL-33 improves synaptic plasticity and memory as AD mice post IL-33 injection show increased LTP and freezing in the fear memory test.Furthermore,IL-33 increases microglia numbers and microglia interaction with plaques.CD68+expression is increased with IL-33 administration,showing increased phagocytic activity,and as a result,the plaque size is decreased.These results are indicative of microglia dysfunction in AD mice,as their proliferation and phagocytic properties are decreased.Increased expression of antiinflammatory genes,Arg1 and Fizz1,is detected post IL-33 administration.These findings indicate that microglia dysfunction contributes to the progression of AD and its associated cognitive decline,and IL-33 can increase microglia phagocytic function while decreasing inflammation in the brain which ultimately improves synaptic plasticity (Fu et al.,2016).
TREM2 in microglia regulates important functions such as autophagy and provides a neuroprotective microglia barrier which regulates amyloid compaction and insulation in AD pathology.Microglia processes can tightly wrap around plaques,acting as a physical barrier to promote the formation of highly compact plaques (Yuan et al.,2016).The TREM2 loss of function disrupts microglia barrier and is a risk factor for the development of AD.The disrupted microglia barrier in haploinsufficient TREM2 AD mice alters plaque packing by increasing filamentous plaques and progressing neuronal loss and disease pathology (Yuan et al.,2016;Meilandt et al.,2020).TREM2 also regulates microglial responses in AD by inhibiting microglia autophagy and sustaining cellular energetic metabolism.Trem2–/–5XFAD transgenic mice show enhanced autophagy,and a similar effect (i.e.an increase in autophagic-like vesicles) is also observed in post-mortem brain sections from R47H and R62H heterozygous AD patients compared to the AD patients homozygous for the common TREM2 variant (Ulland et al.,2017).When Trem2–/–5xFAD mice are treated with the creatine analog 1-carboxymethyl-2-iminoimidazolidine (cyclocreatine),there is a decrease in multivesicular structures in microglia,an increase in microglia number near plaques,and a significant decrease in plaqueassociated neuronal dystrophy in TREM2 deficient mice,indicating that TREM2 contributes to microglial dysfunction in AD pathology by disrupting its metabolism and altering microglia barrier functions (Yuan et al.,2016;Ulland et al.,2017;Meilandt et al.,2020).
While TREM2 deficiency has been indicated in AD progression,a transcriptomic analysis of increasedTrem2dosage in 5xFAD mice revealed differentially expressed subsets of microglia genes between normal 5xFAD and 5xFAD/TREM2 mice,which regulate phagocytic activity by altering their interaction with plaques (Lee et al.,2018).IncreasingTrem2gene dosage changes the morphology of microglia from hypertrophic amoeboid in 5xFAD mice to ramified in 5xFAD/TREM2 mice.In addition to morphological changes,5xFAD/TREM2 mice have increased expression of CD68+microglia indicative of increased phagocytosis,and as a result,improved memory in the contextual fear conditioning test (Lee et al.,2018).Altogether these studies demonstrate a critical role of microglia TREM2 in cognitive deficits in AD.
In addition to IL-33 and TREM2,glucocorticoids (GC) are also involved in AD cognitive deficits via microglia activation,as 3xTg-AD mice injected with GC agonist dexamethasone leads to loss in dendritic spine density in the CA1,while mice treated with GC antagonist mifepristone show an increase in spine density and spatial memory in the Y maze task (Pedrazzoli et al.,2019).
Microglia and traumatic brain injury
As a response to trauma,microglia numbers increase in the brain after TBI,and the active microglia play a negative role in synaptic plasticity and cognition.Importantly,there is a significantly greater number of microglia activated in the aged injured brain in comparison to young,suggesting a synergy effect of aging and TBI on microglia activation(Krukowski et al.,2018;Ritzel et al.,2019).This can be related to an increased expression of C1q in the aged brain 7 to 30 days after brain injury,and thus the increased signaling of the complement cascade (Krukowski et al.,2018).C1q and synapse colocalization is associated with a decline in synapses.Microglia-mediated loss of synapses negatively affects synaptic plasticity,and thus impairs learning and memory in aged injured mice in the novel object recognition.BothC3–/–KO and anti-C1q antibody-treated aged injured mice show improved memory compared to the mice without C3 mutation or C1q antibody treatment,indicating a critical role of C1q and CR3 in microglia-regulated synaptic plasticity and cognitive deficits in aged mice with TBI.
While cognitive impairment is present in the initial stages of neuronal injury,it tends to exist even after recovery(Makinde et al.,2017).A possible cause of this is the change in microglia gene expression after TBI compared to control mice,specifically long-lasting changes in genes associated with synaptic plasticity.Some LTP-associated microglia genes,includingPtpn5,Shank3andSqstmq,are progressively upregulated from day 7 to day 90 following TBI.These genes have also been linked to other neurocognitive impairment disorders such as AD,schizophrenia and autism (Makinde et al.,2020).Ptpn5expression is involved in LTD in different brain areas including the hippocampus.The progressive upregulation ofPtpn5can induce learning and memory deficits by eliminating glutamate receptors on neurons(Makinde et al.,2020).This result suggests that changes in the expression levels of plasticity-related microglia genes can contribute to cognitive deficits that progress after initial TBI recovery.
Microglia and HIV-associated neurocognitive disorder
Microglia dysfunction in HIV patients contributes to HAND which is highly prevalent in HIV-infected patients even with cART treatment.Microglia-mediated chronic inflammation could be a culprit of cognitive deficits such in verbal fluency,executive function,learning and memory,and motor function that are characterized in cognitive phenotypes in HAND (Dastgheyb et al.,2019).HIV-1 transactivator of transcription (Tat) protein has been shown to activate the NOD-like receptor protein 3 (NLRP3) inflammasome pathway in microglia (Chivero et al.,2017;Rawat et al.,2019),which is a complex regulating the formation of proinflammatory cytokine IL-1β from the activation of caspase-1 (He et al.,2019).The activation of NLRP3 happens in two steps:firstly,there is an increase in pro-IL-1β that is termed priming,and then caspase-1 cleaves and activates IL-1β for secretion from microglia (Chivero et al.,2017).This contributes to the downregulation of NLRP3 regulator miR-223,and as a result,leading to Tat-induced microglial inflammasome activation which contributes to HAND.Furthermore,NLRP3 activation in human microglia in the presence of HIV specific GU-rich single-stranded RNA enhances proinflammatory cytokines including IL-1β and IL-18,and neurotoxic cytokines and complement components including TNF-α,IL-1α,and C1q(Rawat et al.,2019).The release of cytokines in activated microglia resulted in significantly increased cytotoxicity and cell death in human primary neurons.Activation of NLRP3 also resulted in mitochondrial damage in microglia,with a loss in integrity and significant increase in reactive oxidative species in part due to disrupted mitophagy (Thangaraj et al.,2018;Rawat et al.,2019).The disruption of mitophagy increases caspase-1 activity which mediates neurodegeneration and cognitive deficits in HIV (Rawat et al.,2019).In a different study,RK-33 which is an inhibitor of Dead Box RNA Helicase 3 (DDX3),was discovered through the analysis of previously unknown targets connected to HAND via an AI-based literature mining system called MOLIERE.RK-33 reduces microglia activation and neuronal death in rodent primary cortical cell cultures treated with Tat (Aksenova et al.,2020),suggesting a potential treatment for Tat-induced toxicity by reducing activated microglia via targeting DDX3.
Microglia and mental disorders
Impaired microglia autophagy during neurodevelopment is implicated in the pathology of autism spectrum disorder which is characterized by social communication impairment and repetitive behaviors (Kim et al.,2017;Lord et al.,2020).To assess the role of microglia in social behavior and dendritic spine density in autism,Atg7fl/fl/Lyz2-Cre mice are designed to knockout an essential autophagy gene,autophagy-related genes (Atg),in microglia.Microglia synaptic pruning is dysfunctional in Atg7fl/fl/Lyz2-Cre mice during neurodevelopment,and reduced pruning results in larger dendritic spines in the primary auditory cortex and secondary somatosensory cortex.In Atg7fl/fl/Lyz2-Cre mice primary microglia culture,microtubule-associated protein 1A/1B-light chain 3-II,a marker of autophagosomes,is decreased (Wang et al.,2013).The autophagy in Atg deficient mice is impaired,resulting in an accumulation of immatures synapses,of which is further validated by increased thickness,fragments,and MAP2-positive dendritic filopodia in the neurons (Kim et al.,2017).Functional connectivity is also altered in Atg7fl/fl/Lyz2-Cre mice as well,as seen via fMRI and global fMRI BOLD signal analysis.Social interaction in Atg7fl/fl/Lyz2-Cre mice is impaired compared to control in the three-chamber social interaction assay,along with a significant increase in repetitive behavior in the marble-burying test (Kim et al.,2017).Altogether,the impaired autophagy in microglia disrupts the neuronal connectivity and causes autistic-like behaviors.
Microglial dysfunction regulates cognitive deficits associated with depression.Learned helplessness (LH) paradigm is a depression model in which mice receive repeated inescapable foot-shocks.Mice subjected to LH have lower dendritic spine density and increased microglia activation.Consequently,these mice show impaired object memory in the NOR and spatial working memory deficits in the Y-maze task.Proinflammatory cytokines and inflammation in the brain are associated with both cognitive impairment and development of depression (Zuckerman et al.,2018),whereas anti-inflammatory cytokine IL-10 expression is associated with increased cognitive function and plasticity (Jung et al.,2019).LH mice have lower microglia IL-10 expression compared to control mice.The administration of IL-10 to LH mice improves learning and memory performance (Worthen et al.,2020).Additionally,the deficits in hippocampal dendritic spine density,NOR and spatial memory are rescued by the ablation of microglia with PLX5622 in mice with LH.These results demonstrate that decreased expression of IL-10 in microglia induces neuroinflammation and plays an important role in the loss of spine density and learning and memory deficits in depression.
Similar to its role in autism and depression,microglia also contribute to cognitive deficits associated with post-traumatic stress disorder (PTSD).A number PTSD studies have shown that there are increased peripheral inflammatory markers and neuroinflammatory markers in PTSD patients as well as increased proinflammatory cytokines,such as TNF-α and IL-1β,in the hippocampus of PTSD mice models (Passos et al.,2015;Zhang et al.,2020b;Li et al.,2021).When electric foot-shocks are used to induce PTSD,microglia are activated in response to the electric foot-shock,and an increase in the number of microglia in the hippocampus and prefrontal cortex is found in the Cx3cr1-GFP mice.In addition to an increase in the number,the microglia show altered morphology including enlarged soma area,reduced branches and reduced arborization (Li et al.,2021).PTSD mice have decreased mushroom and stubby dendritic spine density in the CA1 region of the hippocampus,which is indicative of synaptic dysregulation that may lead to learning and memory impairments.To further investigate the role of microglia in memory deficits in PTSD,genetic or pharmacological microglial deletion using PLX3397 or diphtheria toxin (DT) inCx3cr1CreERmice ameliorates PTSDinduced proinflammatory cytokines gene expression and memory and anxiety deficits (Li et al.,2021),suggesting that the anxiety and memory deficits in PTSD can be attributed to the increased microglia activation and neuroinflammation.
Summary
In this review we first summarized the recent studies of microglia-mediated synaptic pruning.Microglia regulate synaptic structural remodeling by physically contacting neuronal synapses with their motile processes.Microglia regulate synapse elimination in an activity dependent process,as active synapses are selectively maintained while nonessential synapses are eliminated (Filipello et al.,2018;Györffy et al.,2018;Lehrman et al.,2018;Nemes-Baran et al.,2020;Nguyen et al.,2020;Wang et al.,2020).Because of the critical role of microglia in synaptic structural remodeling and their ability to sense active synapses,microglia are able to regulate neuronal activity via a negative feedback mechanism(Badimon et al.,2020) and synaptic tagging and plasticity (e.g.LTP and LTD) partially via the PI3K,BDNF and CREB signaling(Chen et al.,2017;Burk et al.,2018;Raghuraman et al.,2019;Saw et al.,2020).Because microglia regulate synaptic plasticity which is the cellular mechanism of learning and memory(Lisman et al.,2018),several recent studies demonstrate the critical role of microglia in normal learning and memory,including memory strength and quality (Vainchtein et al.,2018;Wang et al.,2020),and in experience-dependent plasticity including the plasticity in the barrel cortex after the removal of whiskers and in the visual cortex after monocular deprivation (Sipe et al.,2016;Wang et al.,2016;Kalambogias et al.,2020).Due to the importance of microglia in synaptic pruning,synaptic plasticity and learning and memory,it is not surprising that abnormal microglia activation and the resulting neuroinflammation have been shown to be a main causal mechanism of the cognitive deficits associated with normal aging and different diseases including AD,TBI,HAND,and mental disorders such as autism,depression and PTSD(Fu et al.,2016;Kim et al.,2017;Ulland et al.,2017;Elmore et al.,2018;Krukowski et al.,2018;Lee et al.,2018;Zöller et al.,2018;Rawat et al.,2019;Li et al.,2020b,2021;Makinde et al.,2020;Meilandt et al.,2020;Nguyen et al.,2020;Worthen et al.,2020).
By reviewing the role of microglia in synaptic plasticity and learning and memory,we want to emphasize that microglia are not only the immune and neuronal support cells of the brain,but also an essential regulator for synaptic structure and memory strength and precision,as well as an important target for the treatment of cognitive impairment with aging,AD and other disorders.Fortunately,currently there are many welldeveloped tools and techniques for microglia study,including microglia inducibleCx3cr1CreERmice,microglia elimination or inhibition drugs such as PLX and Clodronate,the single-cell or single-nucleus RNA sequencing to study microglia specific signaling,and microglia labeling such as the Cx3cr1-GFP mice and two photon imaging to lively monitor the neuronmicroglia interactions (Wang et al.,2018;Zhou et al.,2020;Li et al.,2021).In addition to the currently available tools,the development of a more efficient and convenient viral tool to allow microglia-specific infection and expression should greatly facilitate microglia study.Besides the current literature of microglia role in synaptic plasticity,learning and memory and cognitive deficits,we expect that more future studies of microglia-mediated activity-dependent synaptic tagging and remodeling should further improve our understanding of the role of microglia in cognitive function both under normal and diseased states.
Acknowledgments:We thank Celina Huang for her help to make the figures‚and thank Alexandra Bui‚Roshni Jogin and Zade Zahlan for their suggestions to the review writing.
Author contributions:JC wrote the section of microglia in plasticity and memory deficits.SS and HYH wrote the sections of microglia regulation of snaptic pruning‚plasticity and learning and memory.MZ helped to write and edit all sections.All authors approved the final version of the manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Financial support:None.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal‚and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License‚which allows others to remix‚tweak‚and build upon the work non-commercially‚as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Towards a comprehensive understanding of p75 neurotrophin receptor functions and interactions in the brain
- Stroke recovery enhancing therapies:lessons from recent clinical trials
- Functional and immunological peculiarities of peripheral nerve allografts
- MicroRNA expression in animal models of amyotrophic lateral sclerosis and potential therapeutic approaches
- Significance of mitochondrial activity in neurogenesis and neurodegenerative diseases
- GDNF to the rescue:GDNF delivery effects on motor neurons and nerves,and muscle re-innervation after peripheral nerve injuries