
交联催化剂对聚三唑聚醚弹性体性能的影响
2022-08-13 09:04张永丽杨荣杰张维海高喜飞姜恩周张宇轩
含能材料 2022年8期
张永丽,杨荣杰,张维海,高喜飞,姜恩周,张宇轩
(1. 西安北方惠安化学工业有限公司,陕西 西安 710302;2. 北京理工大学材料学院,北京 100081)
0 引言
硝酸酯增塑的聚醚(NEPE)高能复合推进剂通常采用高氯酸铵(AP)作氧化剂。随着固体推进剂对高能和低特征信号的发展要求,AP 逐渐被新型高能氧化剂二硝酰胺铵(ADN)所取代。然而,由于NEPE 推进剂中的异氰酸酯易与ADN、水等发生反应[1]生成气体产物CO2和NH3,使固体推进剂固化后出现气孔,降低了推进剂的力学性能、安全性、可靠性[2];且异氰酸酯与推进剂中常用硝酸酯增塑剂反应生成的亚硝基化合物[3]也会降低存储过程中推进剂力学性能。因此,需要寻求合成一种对水分不敏感的新型黏合体系的方法,解决异氰酸酯和氧化剂ADN 遇水反应的问题,使ADN 较好的应用于推进剂中,达到高能洁净的目的。叠氮/炔基的点击化学反应固化体系作为一种新颖的非异氰酸酯固化体系[4]因此而受到关注。研究发现,ADN 与点击化学固化体系相容性好[5],ADN 可能被应用于高能微烟推进剂、高能低特征信号固体推进剂中。
点击化学反应是一种1,3-偶极环加成反应,由Huisgen[6]首次提出,叠氮化合物与炔基化合物可以发生1,3-偶极环加成反应生成1,4-二取代和1,5-二取代-1,2,3-三唑环化合物,这类反应的产物是1,4-三唑和1,5-三唑的混合物。近年来,多处报道采用一价铜盐Cu(Ⅰ)作为催化剂可选择性地得到1,4-三唑的产物,这种立构选择性便是典型点击化学反应[7-8]。利用点击化学原理[9-11],Cu(Ⅰ)催化1,3-偶极环加成反应(CuAAC)替代氨基甲酸酯反应,以端炔基聚醚和叠氮化合物组成新的固化体系,不仅具有反应条件温和,可生成高的生成焓的三唑环的特点,而且燃烧时释放出氮气清洁,被广泛用于含能化合物的分子构建,以解决传统固体复合推进剂固化体系存在的问题。
为此,本研究采用端炔基环氧乙烷-四氢呋喃共聚醚(PTPET)作为黏合剂,多官能度叠氮化合物作为固化剂,通过点击化学反应,采用交联催化剂2,4-戊二酸铜-环辛二烯复合物催化固化,生成聚三唑交联弹性体,利用红外光谱(FTIR)、热重分析法(TG)、平衡溶胀法、动态热机械分析法(DMA)等方法研究了不同含量的交联催化剂对聚三唑交联弹性体性能的影响,并对聚三唑交联弹性体热稳定性与常规的PET 弹性体进行对比分析研究。
1 实验部分
1.1 实验原料
端炔基环氧乙烷-四氢呋喃共聚醚(PTPET),北京理工大学材料学院实验室自制,数均分子量Mn约为4000,官能度为2.0,端炔基值为0.467 mmol·g-1。叠氮固化剂为缩水甘油叠氮聚合物(GAP),含氮量为37.2%,数均分子量Mn为480,官能度为3.82,洛阳黎明化工研究院。交联催化剂,2,4-戊二酸铜-环辛二烯复合物,Alfa Aesar 公司。无水乙醇、癸二酸二正辛酯(DOS)、甲苯,分析纯,北京化学试剂公司。
1.2 聚三唑交联弹性体的制备
将不同含量的交联催化剂溶解于DOS 中,配制5 %的透明DOS 溶液;再将黏合剂PTPET 与叠氮固化剂GAP 以固化参数R=1.0 进行混合,加入DOS 溶液中,充分搅拌均匀,置于真空干燥箱,抽真空除去气泡,浇入60 mm×40 mm×3 mm 的聚四氟乙烯模具,50 ℃下固化3 d 制成聚三唑弹性体胶片,记为S1~S4,取出放入干燥器中静置1 周后,进行性能测试。S1~S4 相关参数见表1。
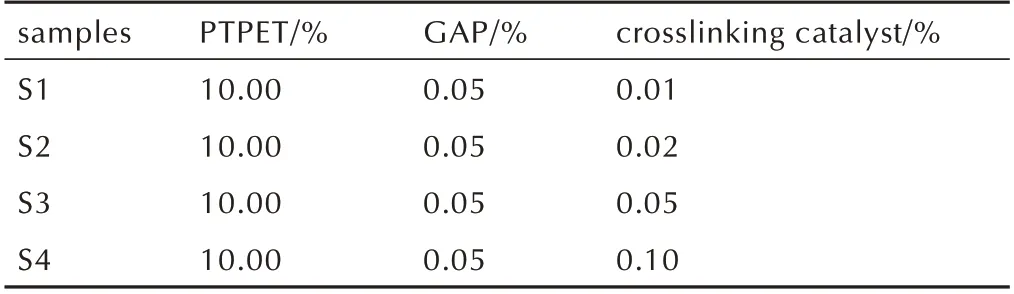
samples S1 S2 S3 S4 PTPET/%10.00 10.00 10.00 10.00 GAP/%0.05 0.05 0.05 0.05 crosslinking catalyst/%0.01 0.02 0.05 0.10
1.3 仪器及测试条件
(1)红外表征:傅里叶变换红外光谱(FTIR),Nicolet 6700,常温测试,扫描次数为32 次。
(2)热稳定性测试:热失重分析仪TG:Netzsch 209 F1,样品量2~3 mg,测试气氛N2,流量50 mL·min-1,加热速率10 K·min-1,加热范围40~500 ℃。
(3)密度测定:METTLER TOLEDO XP204,样品尺寸10 mm×5 mm×2 mm,常温测试,d=0.1 mg。
(4)交联网状结构测定:采用平衡溶胀法[12],取一定体积的样品S1~S4(约0.2 g,10 mm×5 mm×2 mm)浸泡于甲苯中,间隔24 h 后取出用干燥的滤纸擦干表面溶剂,称其质量,直至30 min 内相邻两次质量差小于0.005 g。
根据质量加和性原理[13]采用式(1)计算聚三唑弹性体的体积溶胀率,

式中,ω0为聚三唑弹性体起始质量,g;ω为溶胀后的质 量,g;ρ1为 溶 剂 的 密 度,g·cm-3;ρ2为 弹 性 体 的 密度,g·cm-3。
(5)动态力学性能:动态力学分析仪(METTLER Instrument SDTA861e),样品S1~S4尺寸5 mm×5 mm×2 mm,加 热 速 率2 K·min-1,频 率1 Hz,加 热 范围-100~100 ℃。
(6)力学性能:电子拉力试验机(CMT4101,MTS公司),参考GB/T 258-2009 测试标准[14],样品为标准哑铃状,拉伸速率20 mm·min-1,测试温度20 ℃。
2 结果与讨论
2.1 原材料与聚三唑交联弹性体的红外表征
为研究原材料基团间的反应程度,对黏合剂PTPET、叠氮固化剂GAP、DOS 及聚三唑交联弹性体样品S1~S4 进行了红外表征,其红外谱图如图1 所示。
由图1a 的原料的特征峰可以看出,黏合剂PTPET红外谱图中特征峰3247、664 cm-1分别为端炔基C—H 伸缩振动和弯曲振动吸收峰,2929、2851 cm-1为不同种类亚甲基的伸缩振动吸收峰,1101 cm-1为C—O—C 醚键伸缩振动吸收峰;叠氮固化剂GAP 红外谱图中特征峰3425 cm-1为端—OH 伸缩振动吸收峰,2931,2873,1437cm-1为不同种类亚甲基的伸缩振动和弯曲振动吸收峰,2084 cm-1为叠氮基—N3缩振动吸收峰,1104 cm-1为C—O—C 醚键伸缩振动吸收峰;溶剂DOS 红外谱图中特征峰1738 cm-1为羰基—C=O 的伸缩振动吸收峰。
对比图1a 原材料和图1b 样品S1~S4 的红外谱图可以看出,叠氮固化剂GAP 中的2084 cm-1处—N3吸收峰和黏合剂PTPET 中的3247 cm-1处端炔基C—H吸收峰完全消失,S1~S4 中特征峰2929,2851 cm-1为不同种类亚甲基的伸缩振动吸收峰,1101 cm-1为C—O—C 醚键伸缩振动吸收峰,1738,1642 cm-1处为叠氮基与炔基反应生成的三唑环特征吸收峰[15],表明黏合剂PTPET 与叠氮固化剂GAP 生成了聚三唑交联聚合物。
2.2 聚三唑交联弹性体与PET 弹性体的热稳定性
为研究聚三唑交联弹性体S1~S4 的热稳定性,在N2气氛10 ℃·min-1升温速率下,对样品S1~S4 和传统的PET 弹性体进行热重分析法(TG)测试,得到其质量损失曲线(TG)和质量损失速率曲线(DTG)如图2所示。
由图2a 可以看出,S1~S4 的初始分解温度(T5%)分别为317.1,270.1,243.1,238.0 ℃,初始失重温度逐渐降低,这是由于溶剂DOS 分解导致。图2a 还可以看出S1~S4 网络的热失重并未受到影响。
从图2a 可知,样品S3 和S4 在N2气氛下的TG 曲线表现为明显的2 个分解阶段,DTG 曲线上的2 个失重速率峰也验证了这一现象(图2b)。第一阶段S3 和S4 的分解温度大约在270 ℃,为溶剂DOS 的分解;第二个阶段他们的分解峰温约在405 ℃,为聚三唑弹性体的热分解温度,比PET 弹性体的高约8.9 ℃,说明聚三唑弹性体的稳定性较好。S1~S4 主碳链热分解峰温分别为405.2、402.8、406.6、405.2 ℃,分解温度基本相同,说明交联催化剂的含量不影响聚三唑交联弹性体的热分解。这里需要说明,依据文献[16]报道点击化学中的三唑环的热分解温度约在350 ℃,但在图2中S1~S4 分解曲线不明显,分析认为这是由于405 ℃左右S1~S4 的聚醚链主碳链的分解掩盖了三唑环的分解。
2.3 交联催化剂对聚三唑交联弹性体的动态力学性能的影响
为了考察交联催化剂的含量对聚三唑弹性体玻璃化转变温度(Tg)的影响,对样品S1~S4 进行了DMA 研究,得到损耗因子-温度曲线(图3)及动态力学结果(表2)。其中,工程上将损耗因子tanδ峰值对应的温度作为玻璃化转变温度Tg。
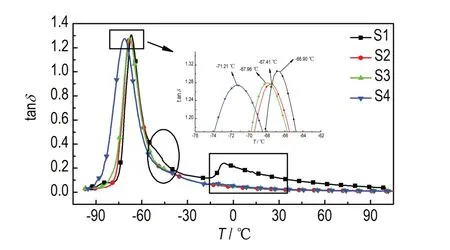
由图3 可以看出,对于交联催化剂含量为0.02%的样品S1 在-45 ℃附近出现了较小的肩峰,在-17~0 ℃附近出现了平台上升现象;当对于交联催化剂含量大于0.02%的样品S2~S4 则未出现肩峰和平台上升现象。分析认为肩峰是因为聚三唑弹性体中存在不规则的未交联的网状结构。未完全交联的样品S1 的网络结构中存在悬挂链,悬挂链具有可以自由自由运动的末端链段,能够通过自由链段的滑移导致了粘弹性松弛,通过粘弹松弛将作用在其上的功能转化为热能,从而引起损耗因子tanδ的增加,出现肩峰。随着交联催化剂含量的增多,样品S2~S4 的悬挂链的数量减少,粘弹性松弛导致的热量损失减少,因此平台上升期后损耗因子tanδ也减少。平台上升期是由于悬挂链的熔融相变导致[14]。样品S2~S4 因网络结构交联完全,无自由移动的链段,不存在肩峰和平台上升现象。

samples S1 S2 S3 S4 tanδ 1.307 1.279 1.277 1.275 Tg / ℃-66.90-67.41-67.96-71.21
从图3 可以看出,样品S1~S4 的玻璃化转变温度Tg随着DOS 含量的增加向低温方向偏移,这是由于聚三唑弹性体中随着DOS 含量的增加,低分子量组分会引起自由体积增大,使PTPET 大分子间的相互作用减弱,链段内旋转势垒减少,活动性增强,从而导致玻璃化转变温度Tg下降[17](表2),交联催化剂含量对聚三唑弹性体的玻璃化温度影响较小。
2.4 交联催化剂对聚三唑交联弹性体的力学性能与网络结构的影响
为了考察交联催化剂含量对聚三唑弹性体的力学性能和网络结构的影响,对样品S1~4 进行了平衡溶胀试验和单轴拉伸试验,得到其溶胀曲线(图4)与单轴拉伸曲线(图5)。
由图4 可以看出,在溶胀初期,样品S1~S4,随着交联催化剂含量的增加,其初始溶胀速率(曲线的斜率)逐渐减小,420 min 左右,溶胀达到平衡。

图4 样品S1~S4 的溶胀曲线Fig.4 Swelling curve of samples S1-S4
图5 是添加不同交联催化剂含量的聚三唑弹性体的常温拉伸曲线。随着交联催化剂含量的增加,样品S1~S4 的拉伸曲线类型无明显变化,无屈服,断裂后能迅速恢复,均表现出一定的高弹性特征。
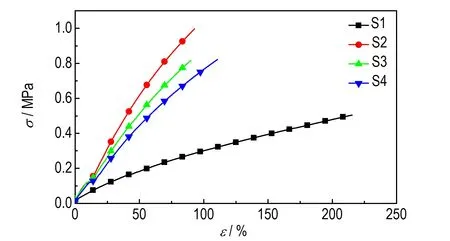
图5 样品S1~S4 的应力-应变曲线Fig.5 Stress-strain curve of samples S1-S4
由图4的结果计算得到聚三唑弹性体S1~S4的交联网络结构参数,结果见表3。其中,弹性体交联网络结构的表观分子量Mc,g·mol-1,可由式(2)Flory-Huggins 理论[18]得到;ν2m,聚合物的体积分数,由式(3)可得;χ1,弹性体和溶剂间的Flory-Huggins 相互作用参数,由Bristow Watson 方程[19](式(4))可得;δp和δs是弹性体和溶剂甲苯的溶度参数,可根据基团加和性[20]计算而得,分别为18.06(J·cm-3)1/2和18.1(J·cm-3)1/2;N0是弹性体交联网络的表观密度,mmol·cm-1,可以根据弹性体的密度和表观分子量Mc(式(5))计算而得。

式中,ρ为弹性体密度,g·cm-3;V为溶剂甲苯的摩尔体积,mL·mol-1;qv为 弹 性 体 溶 胀 平 衡 时 的 体 积 溶胀率。
由表3 和图5 可知,随着交联催化剂含量的增加,样品S1~S4 网络结构中的表观密度(N0)先增加后减小,交联点间的链段平均表观分子量(Mc)先减小后增大,在交联催化剂含量为0.02%时(样品S2)达到极值,平均表观分子量Mc为3483 g·mol-1,表观密度N0为0.301 mmol·cm-1。从表3 数据知,随着交联催化剂含量的增加,样品S1~S4 的最大拉伸强度和弹性模量先增大后减小,断裂延伸率先降低后升高。对于聚三唑弹性体来说,拉伸应变与表观分子量Mc成正比,拉伸强度和模量和表观密度N0成正比,样品S2 的拉伸强度σm为1.00 MPa,断裂延伸率εb为93%,表明其力学性能较优,端炔基与叠氮基反应生成五元聚三唑环刚性硬段,拉伸强度达到最大值,交联网络结构交联完善。聚三唑弹性体S1 具有最大的Mc(9729)和最小的N0(0.109),由于GAP 的叠氮基团与PTPET 的端丙炔基反应不完全,交联网络中存在部分悬挂的侧链[21]。聚 三 唑 弹 性 体S3 和S4 的 表 观 分 子 量Mc和 表观密度N0数值相近,网络结构也交联完全,但由于溶剂DOS 的含量较多,小分子增塑剂增加了链段滑动的柔性,导致拉伸强度降低。总体,样品S2 的交联网络结构完整,力学性能最优。
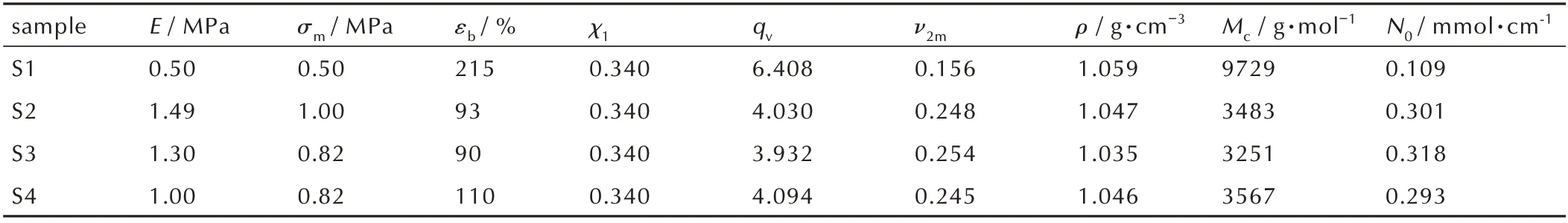
sample S1 S2 S3 S4 N0/ mmol·cm-1 0.109 0.301 0.318 0.293 E / MPa 0.50 1.49 1.30 1.00 σm/ MPa 0.50 1.00 0.82 0.82 εb/ %215 93 90 110 χ1 qv ν2m 0.340 0.340 0.340 0.340 6.408 4.030 3.932 4.094 0.156 0.248 0.254 0.245 ρ / g·cm-3 1.059 1.047 1.035 1.046 Mc / g·mol-1 9729 3483 3251 3567
3 结论
(1)聚三唑交联固体弹性体的热稳定性较PET 弹性体稳定,分解温度约为405 ℃,不随交联催化剂含量的增加而改变。
(2)聚三唑交联固体弹性体S2 网状结构交联完全,且弹性体的弹性模量为1.49 MPa,拉伸强度为1.00 MPa,拉伸断裂延伸率为93%。当增加交联催化剂含量时,对弹性体的交联网络结构影响较小。
(3)聚三唑交联固体弹性体的动态力学性能表明交联催化剂含量较低时交联不完全,易出现损耗因子峰。
(4)交联催化剂溶解于DOS 中,溶解度较小,DOS 含量较多,一定程度上影响弹性体玻璃化转变温度的表征,下一步工作考察快速有效溶解交联催化剂的溶剂。
猜你喜欢
——抗爆炸减压弹性体
橡胶工业(2022年4期)2022-07-19
液晶与显示(2022年2期)2022-03-03
农药学学报(2021年4期)2021-08-26
山西农业科学(2020年11期)2020-11-19
固体火箭技术(2019年4期)2019-09-13
火工品(2019年3期)2019-09-02
福建基础教育研究(2019年10期)2019-05-28
探测与控制学报(2017年3期)2017-07-12
北京理工大学学报(2016年6期)2016-11-22
