
Deciphering the role of PGC-1α in neurological disorders: from mitochondrial dysfunction to synaptic failure
2022-08-08 02:11:08JessicaPanesAlineWendtOscarRamirezMolinaPatricioCastroJorgeFuentealba
中国神经再生研究(英文版) 2022年2期
Jessica D. Panes,Aline Wendt,Oscar Ramirez-Molina,Patricio A. Castro,Jorge Fuentealba,
Abstract The onset and mechanisms underlying neurodegenerative diseases remain uncertain. The main features of neurodegenerative diseases have been related with cellular and molecular events like neuronal loss,mitochondrial dysfunction and aberrant accumulation of misfolded proteins or peptides in specific areas of the brain. The most prevalent neurodegenerative diseases belonging to age-related pathologies are Alzheimer’s disease,Huntington’s disease,Parkinson’s disease and amyotrophic lateral sclerosis. Interestingly,mitochondrial dysfunction has been observed to occur during the early onset of several neuropathological events associated to neurodegenerative diseases. The master regulator of mitochondrial quality control and energetic metabolism is the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α). Additionally,it has been observed that PGC-1α appears to be a key factor in maintaining neuronal survival and synaptic transmission. In fact,PGC-1α downregulation in different brain areas (hippocampus,substantia nigra,cortex,striatum and spinal cord) that occurs in function of neurological damage including oxidative stress,neuronal loss,and motor disorders has been seen in several animal and cellular models of neurodegenerative diseases. Current evidence indicates that PGC-1α upregulation may serve as a potent therapeutic approach against development and progression of neuronal damage. Remarkably,increasing evidence shows that PGC-1α deficient mice have neurodegenerative diseases-like features,as well as neurological abnormalities. Finally,we discuss recent studies showing novel specific PGC-1α isoforms in the central nervous system that appear to exert a key role in the age of onset of neurodegenerative diseases and have a neuroprotective function in the central nervous system,thus opening a new molecular strategy for treatment of neurodegenerative diseases. The purpose of this review is to provide an up-to-date overview of the PGC-1α role in the physiopathology of neurodegenerative diseases,as well as establish the importance of PGC-1α function in synaptic transmission and neuronal survival.
Key Words: Alzheimer’s disease; amyotrophic lateral sclerosis; Huntington’s disease; mitochondrial dysfunction; Parkinson’s disease; PGC-1α; synaptic function; vascular dementia
Introduction
Neurodegenerative diseases (NDs) are a group of neurological disorders characterized by neuronal loss in specific areas of the central nervous system (CNS) and peripheral nervous system (PNS) that can induce the impairment of vital functions like cognitive alterations,uncontrolled motor activities,and all functions that affect daily tasks related with postural balance,movement,memory,language,breathing,cardiac and social behavior (Dugger and Dickson,2017). The more common NDs in the elderly population are Alzheimer’s disease (AD),Huntington’s disease (HD),Parkinson’s disease (PD),amyotrophic lateral sclerosis (ALS) and vascular dementia (VaD). They are characterized by a common pathophysiology feature related with aberrant protein aggregation in the brain,e.g. amyloid beta (Aβ),hyperphosphorylated tau,alpha synuclein (α-syn),huntingtin (HTT) and TAR 43 DNA binding protein (Li et al.,2010; Marsh and Blurton-Jones,2012; Prasad et al.,2019).
Currently,there are no effective therapies for NDs,and the existing treatments are only focused on mitigating the clinical symptoms in patients. The most dramatic example are AD therapies,which have not had any new drugs since 2003 (Cummings et al.,2014). Nowadays,several clinical trials are dedicated to search and find new biomarkers that contribute to the early diagnosis in preclinical or prodromal phases of NDs (Dugger and Dickson,2017; Durães et al.,2018). It has been described that the key mechanisms of excitotoxicity of misfolded proteins are related with fast and progressive processes of oxidative stress,disruption of cell membranes,Ca2+dyshomeostasis,mitochondrial dysfunction,synaptic failure and neuronal death (Li et al.,2016; Dugger and Dickson,2017; Findley et al.,2019).The master transcriptional coactivator PGC-1α (Peroxisome proliferator-activated receptor gamma coactivator 1-alpha) is the main mediator that coordinates mitochondrial biogenesis,cellular respiration,and energy metabolism (Anderson et al.,2008; Bai and Zhang,2016; Jesse et al.,2017). Transcriptional function of PGC-1α is mainly regulated by post-translational modifications (PTMs) of NAD-dependent deacetylase sirtuin-1 (SIRT 1) and 5’AMP-activated protein kinase (AMPK),which act as metabolic sensors increasing ATP production and mitochondrial O2consumption (Anderson et al.,2008; Bai and Zhang,2016).
Analysis of PGC-1α expression patterns show an increase in protein levels in tissues with a high metabolic demand such as brain,heart,skeletal muscle,liver and brown adipose tissue (BAT) in order to promote gene transcription with important consequences in multiple cellular processes such as gluconeogenesis,insulin response,muscle function,glucose metabolism,angiogenesis,contractile function,energy metabolism,and neuroprotection (Lehman et al.,2000; Wenz,2011). Particularly in neuronal survival,it has been observed that PGC-1α also participates in maintaining cholinergic (Zhao et al.,2011; Arnold et al.,2014),glutamatergic (Bartley et al.,2015),dopaminergic (Jiang et al.,2016),and GABAergic synapses in different areas of the CNS and PNS such as the cortex,striatum,substantia nigra,hippocampus,forebrain,spinal cord,and cerebellum (Bartley et al.,2015). Upregulation of PGC-1α has been shown to be neuroprotective against oxidative stress and cell damage by improving mitochondrial function,neuronal maintenance,and protein clearance (St-Pierre et al.,2006; Zolezzi and Inestrosa,2013; Zolezzi et al.,2017). Overall,the aim of this review is to provide a complete picture of the downstream effects associated with the downregulation of PGC-1α in different models as well AD,VaD,HD,PD and ALS thereby providing a new point of view about the putative role of PGC-1α in the maintenance of neuronal function and its role as a potential target for novel pharmacological strategies in the NDs treatments.
Search Strategy and Selection Criteria
Studies cited in this review published from 2000 to 2020 were searched by general medical and science databases (PubMed and Web of Science),using the following keywords “Alzheimer” or “Alzheimer’s disease” “Parkinson” or “Parkinson’s disease”,“Amyotrophic lateral sclerosis”,“Huntington” or “Huntington’s disease”,“Vascular Dementia”,“mitochondrial dysfunction” “PGC-1α isoforms,“brain-specific PGC-1α” for search strategy.
Biological Functions
Canonical PGC-1α and CNS isoforms
PGC-1α is a member of the mammalian family of transcriptional coactivators,along with PGC-1β,and PGC-1-related coactivator (St-Pierre et al.,2003). Initially,PGC-1α was identified as a cold-inducible transcription coactivator of adaptive thermogenesis,but nowadays its well knowing that PGC-1α plays key cellular roles in the regulation of energy metabolism,including gluconeogenesis,insulin sensitivity,mitochondrial biogenesis and ROS detoxification (Puigserver et al.,1998; St-Pierre et al.,2003; Rohas et al.,2007). PGC-1α is a 91 kDa protein encoded by PPARGC1A gene,which spans about 67 kb transcript,including 13 exons located on chromosome 4p15.1 (Esterbauer et al.,1999; Soyal et al.,2012). It has been shown that PGC-1 mRNA expression is markedly elevated upon against a variety of metabolic stimuli e.g.,caloric restriction,hypoxia,exercise,as well high energy demands,in several tissues e.g.,heart,skeletal muscle,kidney,liver,perirenal adipose tissue and brain (Esterbauer et al.,1999; Rohas et al.,2007). Regulation of transcription and gene expression on PPARGC1A gene are a highly regulated by canonical and non-canonical promoters,alternative splicing,as well PTMs e.g.,DNA methylation,acetylation,and phosphorylation (Popov et al.,2017; Soyal et al.,2020).
Increasing evidence have identified novel isoforms of human PPARGC1A transcripts have been identified under control of tissue-specific promoters,such as SNC,liver,BAT,and skeletal (Martínez-Redondo et al.,2015; Soyal et al.,2020). Nowadays,it has been detected in human brain tissue several new brainspecific PGC-1α isoforms under the control of CNS promoter,initiating ~500 kb upstream of canonical promoter (Soyal et al.,2012,2019). However,the differential physiological and pathological roles of these isoforms in the SNC have not completely understood. Particularly,it has been detected a small truncated 17 kDa PGC-1α isoform with uncertain function,some reports suggest that it should be involved in downregulation of full-length PGC-1α ,as well in the onset of some NDs (see below) (Soyal et al.,2019). Actually,it has been suggest that nuclear transcription factors could interact with PGC-1α isoform in the brain to regulated motor function,as well genes related to neurotransmission in the cerebellum,in a sex-dependent brain transcriptional program (Lozoya et al.,2020). Interesting,it has been described that hypoxia,as well polymorphic guanidine thymidine dinucleotide ([GT]n) repeat,can regulated the activation of CNS promoter,in association with several transcription factors e.g.,fork head box protein A2 (FOXA2,and hypoxia-inducible factor 1-alpha (Soyal et al.,2012,2020). Future studies will need to address the mechanisms of regulation between CNS-PGC-1α isoforms,and their specific transcription factors in the brain.
Mechanism of PGC-1α regulation
The mechanisms underlying dynamic modulation of PGC-1α through PTMs have been extensively explored in the past decade. Assembly models suggest that PGC-1α promotes the recruitment of polymerase II,in association with nuclear receptor family transcription factors e.g.,PPARs,ERa,and RXRa,acetyl/methyltransferase complexes,as well several coactivators e.g.,SRC-1,CRE-binding protein,and CBP/p300,and complexes (Wallberg et al.,2003; Aguilo et al.,2016).
It has been described that nuclear/cytosolic sub-localization of PGC-1α are mediated mainly by cytosolic energy sensors; histone acetyltransferase general control non-repressed 5 protein,NAD+-dependent histone deacetylases silent information regulator 2 homolog 1 (SIRT1),and serine/threonine kinase AMPK (Cantó and Auwerx,2009). General control non-repressed 5 protein directly acetylates PGC-1α,and negatively regulates its transcriptional activity,resulting in reduced gluconeogenesis,O2uptake,and mitochondrial biogenesis rates (Lerin et al.,2006; Mutlu and Puigserver,2020). On the other hand,under conditions of energetic stress,NAD+/NADH ratio increases,promoting SIRT1 activation,as well transcriptional activity of PGC-1,mediated by deacetylating at least one of its 13 acetylated lysine residues,resulting in increased expression in antioxidant proteins e.g.,superoxide dismutase (SOD) and glutathione peroxidase (Lehman et al.,2000; Qin et al.,2009). Additionally,AMPK also can modulates PGC-1α/SIRT1 dependent antioxidant program,and mitochondrial homeostasis,in response to disruptions in cellular energy e.g.,cellular adenylate charge and mitochondrial reactive oxygen species (Cantó and Auwerx,2009; Rabinovitch et al.,2017;Figure 1).
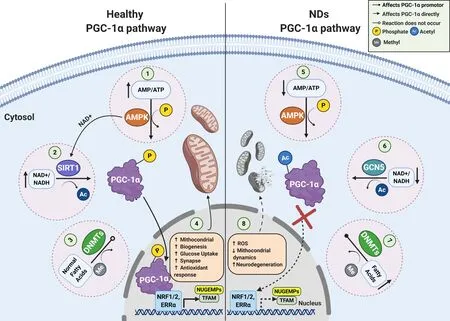
Figure 1|PGC-1α signaling pathways in a normal and pathophysiological context.
Other relevant epigenetic modification that directly affects PGC-1α expression rates is linked to methylation/nucleosome positioning in PGC-1α promoter,which is mediated mainly by DNA methyltransferase (DNMT) isoforms (DNMT1,3A,3B) (Krämer and Handschin,2019; Yang et al.,2020). Interestingly,upregulation in DNA methylation and nucleosome occupancy of PGC-1α promoter,would be implicated in the pathogenesis of some NDs (see below),obesity,diabetes,and liver disease (Su et al.,2015; Narkar,2017; Krämer and Handschin,2019). Elucidation of the mechanisms that regulate PGC-1α and the spatial and temporal identification of all its regulators under conditions of oxidative stress,should uncover promising therapeutic targets for the treatment of metabolic imbalances,and their associated diseases.
PGC-1α and quality control cycle of mitochondria
Quality control cycle of mitochondria depends on several processes that coordinately regulate the number,morphology,integrity of mitochondria. Among them are mitochondrial biogenesis and dynamics,protein turnover,and mitophagy (Celis-Muñoz et al.,2016). Mitochondrial biogenesis depends on the correct performance of mitochondrial/nuclear transcriptional control,which orchestrally coordinates the formation of new functional mitochondria,under several physiological requirements,such as oxidative stress,exercise,synapses,cold,and caloric restriction (Ventura-Clapier et al.,2008; Celis-Muñoz et al.,2016).
It has been described that PGC-1α to reach maximal bioenergetics capacity,acts in association with nuclear respiratory factors,such as nuclear respiratory factors 1 and 2 (NRF-1,NRF-2),as well nuclear hormone receptors,such as estrogen-related receptor alpha (ERRα,NR3B1),peroxisome proliferator-activated receptor gamma (PPAR-γ),and thyroid hormone receptors (Dominy and Puigserver,2013). The most of mitochondrial genes (~1000) are under the control of nuclear transcriptional complexes to raises the transactivation of nuclear genes encoding mitochondrial proteins e.g.,ATP synthase subunits,detoxification proteins,mitochondrial protein import machinery,as well mitochondrial transcription factor A (Schreiber et al.,2004; Rodgers et al.,2008; Celis-Muñoz et al.,2016; Ploumi et al.,2017). Posteriorly into the mitochondrial matrix,mitochondrial transcription factor A binds to the promoter of mitochondrial DNA,and regulates the expression of the rest 13 mitochondrial proteins,coded by circular mitochondrial genome (Gabrielson et al.,2014).
On the other hand,the balance between fusion and fission constitutes a key phenomenon on determining the size,morphology,and distribution of mitochondria. Mitochondrial dynamics are highly coordinated by mitofusin 1,2 (Mfn1,Mfn2),and mitochondrial dynamin like GTPase for cristae fusion,while dynamin-related protein 1 and mitochondrial fission 1 protein have been related with the constriction and fission of mitochondrial cristae (Celis-Muñoz et al.,2016). Actually,several evidences have postulated that mitochondrial dynamics would be affected by mitochondrial biogenesis,in cross-regulatory circuits (Uittenbogaard and Chiaramello,2014; Dorn et al.,2015; Song et al.,2015). PGC-1α-deficient mice showed an altered mitochondrial dynamics,including mitochondrial fragmentation and aberrant elongation,as well defects in mitochondrial fusion/fission proteins,such as Mfn1 and dynamin-related protein 1 (Uittenbogaard and Chiaramello,2014; Dabrowska et al.,2015; Zhang et al.,2020). Improving mitochondrial biogenesis rates may be a potential target for the rectification of diseases associated with mitochondrial morphology defects.
PGC-1α and mitochondrial dysfunction in the CNS
Considering that numerous neuronal mechanisms depends on ATP synthesis and calcium homeostasis,the maintenance of mitochondrial function is essential for key biochemical processes,including Na+/K+ATPase pump,purinergic receptors activity,and neurotransmission (Ly and Verstreken,2006; Dabrowska et al.,2015; Godoy et al.,2019). Furthermore,there is increasing evidence indicated that mitochondrial dysfunction might contribute to the pathogenesis of several diseases,such as diabetes,heart,liver and kidney diseases,loss of muscle coordination,aging and several NDs (Dorn et al.,2015; Cai and Tammineni,2017; Prasad et al.,2019). It has reported that PGC-1α downregulation has associated with mitochondrial dynamics alteration,raises in ROS production,Ca2+dyshomeostasis,oxidative phosphorylation reduction,and ATP production imbalance (Cantó and Auwerx,2009; Dabrowska et al.,2015; Martínez-Redondo et al.,2015;Figure 1).
In vitroandin vivostudies have indicated that PGC-1α regulation would involve in the support of multiple neuronal pathways in the brain,including hippocampus,substantia nigra,cortex,and striatum (Li et al.,2004; Cowell et al.,2007; Uittenbogaard and Chiaramello,2014; Dabrowska et al.,2015). Particularly in neuronal survival,it has been observed that PGC-1α also participates in maintaining cholinergic (Zhao et al.,2011; Arnold et al.,2014),glutamatergic (Bartley et al.,2015),dopaminergic (Jiang et al.,2016),and GABAergic synapses in different areas of the CNS and PNS such as the cortex,striatum,substantia nigra,hippocampus,forebrain,spinal cord,and cerebellum (Bartley et al.,2015). Upregulation of PGC-1α has been shown to be neuroprotective against oxidative stress and cell damage,by improving mitochondrial function,neuroinflammation,protein clearance,and neuronal maintenance (St-Pierre et al.,2006; Zolezzi and Inestrosa,2013; Zolezzi et al.,2017; Rius-Pérez et al.,2020). It is not possible to conclude a complete explanation for the cellular pathways controlled by PGC-1α during excitotoxicity,but selectivein vivoPGC-1α activation could contribute to understanding physiological effects of mitochondrial dysfunction on synaptic network.
Alzheimer’s Disease
The etiology of AD has not been completely determined and the discussion is still open. Cholinergic and glutamatergic dysfunction have been associated with the early cognitive impairments observed mainly in the hippocampus,cortex,and amygdala of AD patients (Castellani et al.,2010; Chen and Mobley,2019). There are two classical histopathological biomarkers detected in AD: intracellular neurofibrillary tangles (Kosik et al.,1986),and extracellular amyloid plaques (Glenner and Wong,1984) triggered by aberrant misfolding and accumulation of hyperphosphorylated tau protein and Aβ,respectively (Glenner and Wong,1984; Kosik et al.,1986; Ballard et al.,2011). Currently,there is a large body of evidence indicating that the soluble oligomers of the Aβ peptide are the most neurotoxic agent because they produce the main aspects of AD pathology,e.g. Ca2+dyshomeostasis and overload,ATP leakage,mitochondrial dysfunction,reticular stress,neuronal loss,synaptic failure and upregulation of the ionotropic channels such as P2XR and NMDAR (Fuentealba et al.,2011; Saez-Orellana et al.,2016,2018; Sinnen et al.,2016; Godoy et al.,2019; Panes et al.,2020).
PGC-1α is a key mediator in the pathogenesis of AD
Increasing evidence postulate that the impairment of the PGC-1α pathway would be associated with the early loss of synaptic maintenance in AD brain regions like the hippocampus and cortex (Cheng et al.,2012; Bartley et al.,2015; Dong et al.,2020; Singulani et al.,2020;Additional Table 1). In fact,it has been observed in several neuronal Aβ toxicity models and patients with moderate and severe clinical dementia rating,that PGC-1α downregulation is critical for the early onset of the disease and for the defective responses in neuronal circuits of AD patients (Qin et al.,2009; Pedrós et al.,2014; Bartley et al.,2015; Dong et al.,2020; Terada et al.,2020). Additionally,it was shown that the level of PGC-1α markedly decreases in correlation with diminished mitochondrial functionality and nonamyloidogenic processing of APP in Tg2576 AD and APP/PS1 mice (Qin et al.,2009; Pedrós et al.,2014; Dong et al.,2020). Recently,it was found that soluble oligomers of the Aβ peptide were able to induce a loss of the interaction between PGC-1α and SIRT1 by decreasing its nuclear translocation and potentiating the imbalance in mitochondrial dynamics (Panes et al.,2020).
Studies in different AD transgenic models have demonstrated that the upregulation or overexpression of PGC-1α was able to mitigate neurodegeneration,Aβ accumulation,and cognitive failure observed in these models (Katsouri et al.,2016; Dong et al.,2020). Effectively,injection of the lentiviral vector human PGC-1α into the hippocampus and cortex areas of APP23 transgenic animals induced a decrease in Aβ production,pyramidal neurodegeneration,impairment of spatial memory,β-secretase transcription,microglial activation,downregulation of neurotrophic factors and the formation of amyloid plaques suggesting an important role of PGC-1α for the control of toxic Aβ species formation (Katsouri et al.,2016). Furthermore,APP/PS1 transgenic mice treated with resveratrol (RSV),a positive modulator of the SIRT1/PGC-1α pathway,showed an improvement in cognitive deficits,learning ability,and spatial memory (Dong et al.,2020). These studies suggest that PGC-1α could have a neuroprotective effect against neuronal loss and synaptic failure observed in AD.
Role of PGC-1α in inhibition/excitation balance in a hippocampal circuit
To understand how PGC-1α can affect synaptic function,it is necessary to evaluate the role of PGC-1α in the maintenance of the inhibition/excitation (I/E) balance in a hippocampal circuit (Cowell et al.,2007; Bartley et al.,2015; Kwakowsky et al.,2018). PGC-1α is highly expressed in GABAergic cell populations in the brain and participates in genetic transcription of the Ca2+buffer protein parvalbumin in forebrain regions such as the cortex,hippocampus,and striatum (Cowell et al.,2007). Particularly,dysfunction in the GABAergic response (e.g. decrease in GABA currents,downregulation in GABA receptors,and the I/E imbalance) have been implicated in the onset of cognitive dysfunction observed during the early stages of AD,mainly in the hippocampus,superior temporal gyrus,and entorhinal cortex (Bartley et al.,2015; Kwakowsky et al.,2018). Studies in pyramidal CA1 cells of PGC-1α knockout (KO) mice demonstrated an increase in frequency-dependent I/E balance of the synaptic network and impaired nest building of the hippocampus mediated by an increase in the amplitude of inhibitory postsynaptic currents in response to Schaffer collateral stimulation in slices from young adult mice. In fact,PGC-1α KO young adult mice had cognitive alterations associated with an increase in inhibition of GABAergic interneurons of the parvalbumin,triggering a circuit dysfunction in CA1 activation and increasing hippocampal kainate-induced gamma oscillations and behavioral impairments (Cowell et al.,2007; Bartley et al.,2015; Kwakowsky et al.,2018;Figure 2).
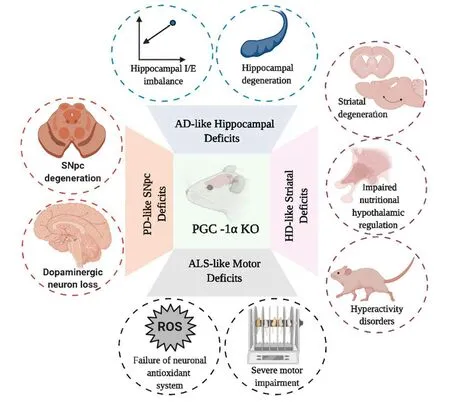
Figure 2|Schematic representation of cellular and behavioral changes observed in several models of PGC-1α KO mice.
Vascular Dementia
VaD is characterized by several neuronal alterations like vascular cognitive deterioration causing a decline in learning,thinking,reasoning and language that is mediated by a progressive hypoperfusion of the cerebral tissues usually seen in patients with heart diseases or cerebral amyloid angiopathy and who have other vascular risk factors such as arterial hypertension,high cholesterol levels,cerebrovascular accidents,diabetes and heart diseases (O’Brien and Thomas,2015). Clinical diagnosis of VaD is based on neuroimaging criteria such as lacunar strokes,white matter lesions,interruption of the blood-brain barrier,neuroinflammation and endothelial dysfunction of brain structures (Venkat et al.,2015; Cipollini et al.,2019). Traditionally,classic VaD treatments that are used are cholinesterase inhibitors,memantine,galantamine or antihypertensive approaches. Unfortunately,the development of specific treatments for VaD has been hampered by the lack of molecular targets or suitable animal models that can emulate the main features of the disease (Baskys and Hou,2007; Gorelick et al.,2011). Currently,the early biomarkers that are being studied in patients with VaD are microRNAs (miRNAs),lipocalin 2 and lipid biomarkers such as high-density lipoprotein and lowdensity lipoprotein (Prabhakar et al.,2017; Cipollini et al.,2019; Llorens et al.,2020).
Emerging evidence of PGC-1α function in VaD
Increasingin vitroandin vivostudies have shown that some compounds or extracts such as RSV,allopregnanolone,Ginkgo biloba extract,and phytoestrogens have an antioxidant effect and enhance metabolic status in VaD patients (Prabhakar et al.,2017; Cipollini et al.,2019). However,there is little data about molecular mechanisms associated with VaD and mitochondrial dysfunction. A recent study in a VaD mouse model induced by bilateral common carotid artery stenosis (BCAS) showed a downregulation in PGC-1α in the CA1 and CA3 area of the hippocampus and a subsequent decrease in the expression of mitochondrial antioxidant genes in the BCAS group (Han et al.,2020;Additional Table 1). Furthermore,the generation of an animal model that overexpresses neuronspecific PGC-1α (nPGC-1α) after BCAS surgery showed an improvement in cognitive function recovery and LTP assessment induced by theta burst stimulation,and also a mitigation in cognitive deficits for learning and memory was observed in the BCAS animals. Additionally,nPGC-1α was able to induce an overexpression of antioxidant mitochondrial genes (e.g.SOD2,Prx3,GPx1,UCP2-5) in the hippocampus after BCAS surgery (Han et al.,2020). This study suggests that PGC-1α could be a potential neuroprotective agent against oxidative stress,energy metabolism imbalance,and neuroinflammation; however,more studies are needed to determine the real impact of PGC-1α function in the synaptic network.
Huntington’s Disease
HD is a progressive and irreversible NDs characterized by progressive impairment in cognitive functions (e.g. memory,executive and visuospatial functions),motor functions (e.g. rigidity,chorea,dyskinesia),psychiatric disorders (e.g. irritability,emotional disorders,apathy and psychosis),and metabolic abnormalities (e.g. wasting and altered energy expenditure) (Wilson et al.,2017). HD etiology is associated with an autosomal dominant trait that appears mainly between 30 to 40 years of age triggered by a codon polymorphism expansion of a glutamine residue (CAG) encoded by the huntingtin gene (HTT,also called IT15) located on chromosome 4p16.3 (The Huntington’s Disease Collaborative Research Group.,1993; Wilson et al.,2017). Mutant HTT (HTTmut) is able to form intracellular aggregates that can interfere with cellular processes that control gene regulation of energy homeostasis and it can induce a dysregulation in corticoestratial connectivity (Cui et al.,2006; Hervás-Corpión et al.,2018),This has been associated to an overactivity of glutamatergic neurotransmission,a reduction in GABAergic output in basal ganglia,excitotoxic cell injury,progressive neurodegeneration and atrophy of specific areas like the amygdala and striatum which increase thalamocortical activity leading to involuntary movements and motor disorders characteristic of the disease (Wilson et al.,2017).
Contribution of PGC-1α in the molecular pathogenesis of HD
Recently,evidence has shown that neurological and psychiatric disorders in HD patients are mediated by a progressive oxidative stress and genetic transcriptional dysregulation of mitochondrial and ribosomal biogenesis against HTTmuttoxicity (Cui et al.,2006; Tsunemi and La Spada,2012; Johri et al.,2013; Jesse et al.,2017). In this context,it has been reported that HTTmutcan induce transcriptional downregulation of PGC-1α,induce an inactivation of the mitochondrial biogenesis process,and disrupt mitochondrial function (Cui et al.,2006; Soyal et al.,2012; Jesse et al.,2017; Jodeiri Farshbaf and Ghaedi,2017). Several animal HD models and HD patients (striatal tissues) have shown that neurodegeneration induced by HTTmuttoxicity was mediated by transcriptional dysregulation of PGC-1α (Additional Table 1) (Cui et al.,2006; Chaturvedi et al.,2009; La Spada,2012; Tsunemi et al.,2012; Intihar et al.,2019). Interestingly,It was suggested that the mechanism responsible for PGC-1α downregulation in striatal neurons in different HD models would be associated to direct interaction and blocking of HTTmutwith the transcriptional activator of PGC-1α CRE-binding protein/TAF4,triggering the behavioral,metabolic,and neuronal disturbances observed in HD (Cui et al.,2006; Chaturvedi et al.,2009). Remarkably,several PGC-1α KO models can emulate the main characteristics of HD,i.e. defective mitochondrial bioenergetics,decreased neurite growth,striatal neurodegeneration,hyperactivity,astrogliosis,impairment in oxidative metabolism,muscle dysfunction,disturbed ribosomal transcription,and a spongiform pattern of lesions in white matter (Johri et al.,2013; Jesse et al.,2017;Figure 2). The nPGC-1α KO model also showed degenerative lesions in the striatum and deep cortical layers,alterations in axonal integrity,and decreases in metabolic programming of hypothalamic neuronal circuits (Ma et al.,2010). The breeding of PGC-1α KO mice with HD KI mice having 140 CAG repeats showed a significant potentiation in stromal neurodegeneration and motor abnormalities in the medial septal nucleus and striatum when compared to HD mice (Cui et al.,2006). Additionally,lentiviral overexpression of PGC-1a in the striatum of R6/2 HD transgenic mice could prevent the neuronal atrophy and dysregulation of axonal transport seen in HD mice (Cui et al.,2006). The breeding of N171-82Q HD transgenic mice with an inducible TRE-PGC-1α mice model was able to decrease aberrant aggregation and turnover of HTTmut,mitigate striatal neurodegeneration,and improve mitochondrial dysfunction observed in HD mice (Tsunemi et al.,2012).
PGC-1α isoforms are associated with early-onset in HD
Interestingly,it has been postulated that “loss of function” of several single nucleotide polymorphism (SNPs) mutations in the PPARGC1A locus that codes for several brain-specific isoforms upstream of exon 2 would be associated with a delay in age of onset of motor symptoms in patients with HD (Weydt et al.,2009; Soyal et al.,2012,2019). In fact,the MAPS genome scan from HD patients showed several SNPs located in the PPARGC1A haplotypes at chromosomal region 4p16-4p15; rs17592631,rs2048025 and rs11737023,that would be associated with a “protective” genotype for HD incidence (Weydt et al.,2009). Furthermore,5′-RLM-RACE experiments demonstrated a novel CNS-promoter for PPARGC1A transcripts that code for several dominant brain-specific PGC-1α isoforms that were able to interact with histone acetyltransferase complexes and dominant negative isoforms (Soyal et al.,2012). Moreover,it has been postulated that these truncated isoforms were able to block transcriptional regulation of the nPGC-1α isoform (Soyal et al.,2019). These novel transcripts should be further studied as a function of their interaction domains with mitochondrial cofactors,and we suggest that PGC-1α isoforms could have a protective effect improving neuronal energy imbalance and neurodegeneration.
Parkinson’s Disease
PD is a progressive NDs characterized by a gradual loss and degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc),which leads to movement and motor disturbances (e.g. tremor at rest,muscle stiffness,bradykinesia,and postural instability) (Emamzadeh and Surguchov,2018; Kouli et al.,2018),and non-motor symptoms (e.g. depression,sleep disturbances,hyposmia,hallucinations,and dementia) (Martinez-Martin et al.,2011). The conventional histopathological biomarker detected in PD patients is composed of abnormal deposits of α-syn aggregates. The majority of abnormally deposited α-syn aggregates are found in the nigral dopaminergic system,particularly intracellularly in the cell soma and also in the neurological processes,also called Lewy and neurites bodies,respectively,that frequently appear between 55 and 70 years of age (Kalia and Lang,2015). PD etiology is associated with several risk factors such as environmental factors (e.g. heavy metals,the mitochondrial neurotoxin MPTP,antipsychotic drugs,and antihemetics),family history (e.g. mutations in SNCA,PARK2,PINK1,PARK7 and PARK8 genes),and other NDs such as AD and HD (Emamzadeh and Surguchov,2018).
Neuroprotective role of PGC-1α in PD progression
Current treatments for PD have been focused primarily on increasing dopamine levels by decreasing oxidative metabolism of dopamine using levodopa,l-DOPA,l-3,4 dihydroxyphenylalanine,dopamine receptor agonists (e.g. amantadine,apomorphine),and also by using mitochondrial monoamine oxidase B (MAO-B) inhibitors (e.g. selegiline and rasagiline) (Emamzadeh and Surguchov,2018; Kouli et al.,2018). Recently,it has been postulated that one of the mechanisms that could be involved with the onset of PD may be associated with a downregulation of the PGC-1α pathway and decrease of mitochondrial metabolism in PD pathogenesis (Zheng et al.,2010; Youdim and Oh,2013; Ciron et al.,2015). Indeed,it has been shown that using either MAO inhibitors or PGC-1α/SIRT1 activators (e.g. M30,HLA-20,LSN and RSV) improves mitochondrial and motor function and also provides neuroprotective effects against oxidative stress and mitochondrial toxins in several models of PD (Youdim,2013; Ferretta et al.,2014; Emamzadeh and Surguchov,2018; Kouli et al.,2018).
In fact,studies have demonstrated the participation of PGC-1α in neuronal survival of SNpc against mitochondrial neurotoxins associated with PD etiology (St-Pierre et al.,2006; Ciron et al.,2015; Stevens et al.,2015; Jiang et al.,2016; Ye et al.,2017). Using RNA interference to study protein function of PGC-1α in SH-SY5Y cells treated with N-methyl-4-phenylpyridinium ion (MPP+),it was found that PGC-1α ablation exerted a potentiation of mitochondrial dysfunction and cell death in MPP+-induced cells (Ye et al.,2017). The generation of a PGC-1α KO model showed a more sensitive phenotype in substantia nigra and hippocampus against oxidative stressors such as MPTP and kainic acid and had decreased axonal integrity and synaptic network integrity in deep cortical layers and striatum,as well as potentiating striatal lesions and oxidative damage (St-Pierre et al.,2006;Figure 2). α-syn overexpression in SNpc in the PGC-1α KO model also potentiated neurodegeneration,altered mitochondrial ultrastructure,and increased oxidative stress; alterations which were corrected by reestablishing PGC-1α expression (Ciron et al.,2015). Furthermore,conditional PGC-1α KO animals showed a progressive loss of dopaminergic neurons in SNpc,an important decrease in dopamine in the striatum,and subsequent reduction in mitochondrial biogenesis (Jiang et al.,2016). Furthermore,several mutations in the parkin and pink proteins also have been associated with the early loss of mitochondrial quality control and oxidative stress processes detected in PD (Matsuda et al.,2010; Stevens et al.,2015; Ge et al.,2020). In adult conditional parkin KO mice,as well as in PD patients,it was found that mitochondrial dysfunction and dopaminergic loss was associated with PGC-1α downregulation in a PARISdependent pathway (Pacelli et al.,2011; Stevens et al.,2015).
Differential role of PGC-1α in dopaminergic neuron survival
Recent studies have revealed that oxidative stress and neurodegeneration of dopaminergic neurons detected in PD models are implicated with several alterations in signaling pathways mediated by PGC1-α regulation (St-Pierre et al.,2006; Siddiqui et al.,2012; Corona and Duchen,2015; Su et al.,2015; Soyal et al.,2019;Additional Table 1). In cortical neurons of MAO- B transgenic mice,and brain tissues of PD patients,it was shown that α-syn is able to bind with the PGC-1α promoter and inhibit its activity in the nucleus,triggering a PGC-1α downregulation,mitochondrial dysfunction,and cell degeneration (Zheng et al.,2010; Siddiqui et al.,2012). Additionally,in substantia nigra and peripheral blood samples from PD patients,studies found that PPARGC1A downregulation and PD clinical features were directly associated with the increase in non-canonical cytosine hypermethylation of the PGC-1α promoter,and in several CpG sites on the PPARGC1A locus (Su et al.,2015; Yang et al.,2020). Recently,it has been suggested that changes on the expression of CNS-specific PGC-1α (CNS-PGC-1α) full length has an important impact on CNS function,while the overexpression of truncated short isoforms in SNpc,would have deleterious effects related with the age of onset of PD (Yang et al.,2018,2020; Soyal et al.,2019). Studies in postmortem tissues from SNpc and globus pallidus of PD patients,indicated that downregulation in full-length PGC-1α isoforms and mitochondrial proteins decreased are associated with dopaminergic neuronal loss and progressive brain disorders observed in PD patients (Weydt et al.,2009; Jiang et al.,2016; Soyal et al.,2019). Taken together,the develop of specific promotor or enhancer of PGC-1a full length expression could be an innovative strategy to improve mitochondrial function for slow down the dopaminergic neurodegeneration in Parkinsonism.
Amyotrophic Lateral Sclerosis
ALS is a motor neuron disorder characterized by a selective loss of motor neurons in the brain,brainstem and spinal cord that leads to a progressive degeneration in upper and lower motor neurons in corticospinal tracts and induces a gradual deficiency in neuromuscular control (e.g. clonus,atrophy,fasciculation,muscle weakness,language problems,respiratory failure and paralysis),which ultimately leads to early death in patients 2-3 years after diagnosis (Muller et al.,2017). Multiparametric neuroimaging studies of ALS patients suggest that the selective anatomical vulnerability pattern in grey matter structures of basal ganglia and commissural white matter tracts could be associated with aberrant accumulation of SOD1 and TARDBP proteins (Bede et al.,2016; Prasad et al.,2019). Older age,gender difference,environment,oxidative stress,disturbances of energy metabolism,and a family history of ALS (e.g. C9ORF72,FUS / TLS,TBK,SOD1 and TARDBP gene mutations) have been recognized as risk factors for ALS onset (Tao and Wu,2017).
Antioxidant and neuroprotective role of PGC-1α in amyotrophic lateral sclerosis
Increasing evidence in ALS models have shown that oxidative stress and mitochondrial abnormalities in the motor neurons could be associated with an increase on neuronal excitability and Ca2+dysregulation (Wen et al.,2013; Ragagnin et al.,2019). It has been identified that PGC-1α also participates in: the regulation of gene expression at the neuromuscular junction,increased antioxidant response and increased mitochondrial biogenesis (Wiedemann et al.,1998; Arnold et al.,2014). Interestingly,a downregulation of PPARGC1A expression,and a subsequent decrease in a key mitochondrial genes (e.g. NRF-1,NRF-2,and Tfam),as well changes in the expression of acetylcholine receptor in skeletal muscle tissue were observed in ALS animal models and ALS patients (Eschbach et al.,2013; Ladd et al.,2014; Bayer et al.,2017;Additional Table 1).
Additionally,overexpression of PGC-1α in the SOD1G93A ALS mice model (TgSOD1G93A/PGC-1α) was able to mitigate neuronal death,motor dysfunction,metabolic disturbances and mitochondrial dysfunction in the ventral horn of the lumbar spinal cord of ALS mice (Zhao et al.,2011). Furthermore,breeding transgenic mice with constitutive PGC-1α overexpression in skeletal muscle (MCK-PGC-1α) with transgenic SOD1G37R mice (SOD1G37R/MCK-PGC-1α) was able to improve muscle atrophy,motor activity,mitochondrial dysfunction,and reduced muscle degeneration in SOD1G37R mice; however,it was not able to improve ALS progression or cell survival (Da Cruz et al.,2012; Johri and Beal,2012). Remarkably,PGC-1α KO transgenic mice showed progressive alterations in motor coordination and downregulation of mRNA levels associated with antioxidant and neuroprotective responses (e.g. Tfeb,Sod3 and Bdnf) (Lucas et al.,2012; Bayer et al.,2017;Figure 2).
Emerging evidence of differential expression of PGC-1α in amyotrophic lateral sclerosis tissues
A biphasic tissue-specific PGC-1α expression has been recently demonstrated in ALS patients,SOD1G93A,and a Fus∆NLS/∆NLSmice models revealing that PPARGC1A mRNA levels was upregulated in primary brown adipocytes,skeletal muscle and brown fat,whereas non-canonical isoforms of PGC-1α was downregulated in the spinal cord and brainstem of ALSaffected neuronal tissues (Bayer et al.,2017). Interestingly,down-regulation of CNS-PGC-1α expression and mitochondrial PGC-1α-dependent genes (e.g. Tfeb,Sod3 and Bdnf) were reestablished after lactate stimulation (Bayer et al.,2017). Moreover,it has been observed that CNS-PGC-1α isoforms could also exert a key role in the age of onset and gender differences in the incidence of ALS (Eschbach et al.,2013; Pasquinelli et al.,2016). In fact,downregulation of CNGPARGC1A was more associated in the incidence of male onset of ALS-transgenic mice compared with female ALStransgenic mice. A similar effect was observed in human ALS (Eschbach et al.,2013). Additionally,an interesting study in ALS patients strongly suggested that antioxidant defense impairment and motor atrophy during intensive training were related with Gly482Ser SNP located in the PPARGC1A genomic locus (Pasquinelli et al.,2016). Further studies about the neuroprotective effect of CNS-PGC-1α isoforms and interacting partners of DNA-binding proteins will be essential for understanding the transcriptional role of PGC-1α in the pathogenesis of ALS onset.
Conclusions
Taken together,the group of NDs reviewed and discussed in this work have been diagnosed traditionally by clinical symptoms; in addition,current pharmacological therapies have failed to mitigate neurodegeneration and anatomic disturbances observed in NDs. Here,we reviewed clear evidence supporting the neuroprotective role of PGC-1α against synaptic failure and neuronal death as a key pathway for the onset of these NDs. Downregulation of PGC-1α is key to understanding the toxicity mechanisms of misfolded proteins that would precede the oxidative stress and progressive neuronal death in specific areas of the brain (Tsunemi and La Spada,2012; Bayer et al.,2017; Panes et al.,2020; Singulani et al.,2020). Growing knowledge about immunology and molecular biology strategies to develop humanized antibodies that can treat different types of pathologies,associated to the discussed evidence of the key role of PGC-1α isoforms,could be an essential starting point to open new chemical,or especially biological drug therapeutic strategies focused on improving mitochondrial and neuronal function in patients with NDs.
Author contributions:JDP reviewed the literature and wrote the manuscript. ORM,PAC and AW reviewed the manuscript. JDP and JF designed and reviewed the manuscript. All authors approved the final version of the manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Financial support:This work was supported by Fondecyt 1200908 (to JF) and by the Conicyt 21141247 doctoral grant (to JDP).
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Xu-Qiao Chen,University of California,USA.
Additional files:
Additional Table 1:Summary of the main effects of PGC-1α regulation in different neurodegenerative disease models.
Additional file 1:Open peer review report 1.
Retraction
Retraction: Emerging concepts underlying selective neuromuscular dysfunction in infantile-onset spinal muscular atrophy
https://doi.org/10.4103/1673-5374.314116
The Editors ofNeural Regeneration Researchhave retracted the publication entitled “Emerging concepts underlying selective neuromuscular dysfunction in infantileonset spinal muscular atrophy” (Gollapalli et al.,2021; doi: 10.4103/1673-5374.308073) following concerns stemming from misquoting and,accordingly,faulty characterization of key literature cited.
Based onNeural Regeneration ResearchPolicy on Article Retraction (http://www.nrronline.org/contributors.asp).
Reference
Gollapalli K,Kim JK,Monani UR (2021) Emerging concepts underlying selective neuromuscular dysfunction in infantile-onset spinal muscular atrophy. Neural Regen Res 16(10):1978-1984.

- 中国神经再生研究(英文版)的其它文章
- A Drosophila perspective on retina functions and dysfunctions
- Celeboxib-mediated neuroprotection in focal cerebral ischemia: an interplay between unfolded protein response and inflammation
- Pramipexole,a dopamine D3/D2 receptor-preferring agonist,attenuates reserpine-induced fibromyalgia-like model in mice
- Effects of delayed repair of peripheral nerve injury on the spatial distribution of motor endplates in target muscle
- Neurorehabilitation using a voluntary driven exoskeletal robot improves trunk function in patients with chronic spinal cord injury: a single-arm study
- Gene and protein expression profiles of olfactory ensheathing cells from olfactory bulb versus olfactory mucosa