
缺氧对巩膜成纤维细胞内质网应激反应的激活作用及其对巩膜重塑的影响△
2022-08-07 13:57唐晓兰杨倩颖
眼科新进展 2022年7期
唐晓兰 刘 玲 杨倩颖 李 华
近年来,近视患病率逐年上升,已成为世界范围内的一个公共卫生问题[1]。大量研究表明,近视的发生发展是巩膜重塑的结果。在巩膜重塑过程中,胶原合成减少、降解增加,最终导致巩膜变薄和眼轴增长[2]。研究表明,巩膜缺氧是导致巩膜重塑的始动因素,在缺氧环境中,巩膜胶原蛋白合成减少[3]。此外,已有大量研究证明,组织缺血缺氧可导致内质网应激,但是巩膜缺氧是否可引起内质网应激,以及内质网应激在巩膜重塑中的作用尚未见确切报道。本研究采用体外培养的人胚胎眼巩膜成纤维细胞(HFSF)缺氧模型,模拟近视巩膜缺氧环境,探讨缺氧对巩膜成纤维细胞内质网应激反应的激活作用及其对巩膜重塑的影响。
1 材料与方法
1.1 材料
1.1.1 实验细胞及分组HFSF购自上海冠导生物工程有限公司。取细胞随机分为缺氧0 h组、缺氧12 h组、缺氧48 h组。
1.1.2 试剂与仪器DMEM高糖培养基(美国Gibco公司),胎牛血清(以色列BI公司),流式细胞仪(美国BECKMAN公司),酶标仪(美国Thermo Labsystem公司),CCK-8试剂盒(日本同仁化学研究所),Annexin V-FITC/PI凋亡试剂盒(美国BD公司),化学发光液(美国Millipore公司),FITC 标记的山羊抗兔二抗(北京中杉金桥生物技术有限公司),免疫荧光显微镜(日本Olympus公司),COL1A1、基质金属蛋白酶-2(MMP-2)、肌醇需求酶l (IRE1α)、α平滑肌肌动蛋白(α-SMA)、β-actin、BAX和BCL-2一抗及山羊抗兔IgG二抗均购自英国Abcam公司,P-IRE1α购自美国CST公司。
1.2 方法
1.2.1 细胞培养及缺氧处理将HFSF采用常规DMEM高糖培养基(含体积分数10%的胎牛血清)培养,传代于6 cm培养皿,在37 ℃培养箱中培养24 h后,缺氧0 h组细胞正常培养,缺氧12 h组及48 h组细胞分别在含氧体积分数2%的三气培养箱中缺氧处理12 h及48 h。
1.2.2 Western blot 检测各组细胞IRE1α、P-IRE1α、COL1A1、MMP-2、α-SMA、BAX及BCL-2蛋白表达收集各实验组细胞,提取蛋白后按照BCA试剂盒说明书进行蛋白浓度测定。取样品行SDS-PAGE电泳后转移至 PVDF膜,用50 g·L-1脱脂奶粉封闭1 h后,与相应的一抗 (稀释度均为11000) 4 ℃孵育过夜。TBST洗3次后,与辣根过氧化物酶标记的山羊抗兔二级抗体(15000)共同孵育1 h,TBST洗3次后进行化学发光显影。使用ImageJ软件计算条带的灰度值,以内参为基数,取目的条带灰度值与内参灰度值之比为蛋白相对表达量,其中缺氧0 h组设为1。
1.2.3 免疫荧光染色检测各组HFSF中α-SMA蛋白含量取细胞给予相应时间缺氧处理后用PBS洗3次,使用免疫荧光固定液固定10 min,PBS洗3次;使用Triton X-100通透15 min,PBS洗3次;使用免疫荧光封闭液封闭30 min后加入相应一抗 (α-SMA,1300)4 ℃过夜;PBS洗3次后加入荧光二抗(150)室温避光孵育1 h,PBS洗3次;使用DAPI染核5 min,PBS洗3次;使用抗荧光淬灭封片剂封片后通过Olympus倒置荧光显微镜采集图像。
1.2.4 流式细胞仪检测各组细胞凋亡情况将细胞传代于6 cm培养皿,按实验设计进行相应处理。使用不含EDTA胰蛋白酶消化收集细胞,1200 r·min-1离心5 min。使用PBS洗涤细胞后1200 r·min-1离心5 min,加入100 μL 1 × Buffer重悬细胞,分别加入5 μL PI及Annexin V-FITC并混合均匀,室温孵育15 min,上机前加入400 μL 1×Buffer混匀,使用CytoFLEX流式细胞仪进行检测,CytExpert进行数据分析。
1.2.5 CCK-8检测各组细胞增殖将细胞以每孔4400个接种于96孔板,分别予以相应缺氧时间处理后每孔加入10 μL CCK-8试剂,37 ℃孵育2 h,使用酶标仪测450 nm波长下的光密度(D450),计算各组细胞增殖情况。
1.3 统计学分析使用GraphPad Prism 8进行统计分析,计量资料以均数±标准差表示。多组间比较采用单因素方差分析。检验水准:α=0.05。
2 结果
2.1 各组细胞IRE1α、P-IRE1α、COL1A1、MMP-2、α-SMA、BAX及BCL-2蛋白表达情况Western blot检测结果显示:与缺氧0 h组相比,缺氧12 h组细胞IRE1α、α-SMA蛋白表达均升高,COL1A1蛋白表达降低,差异均有统计学意义(均为P<0.05),其余蛋白表达变化不明显,差异均无统计学意义(均为P>0.05);与缺氧0 h组相比,缺氧48 h组细胞IRE1α、P-IRE1α、MMP-2、α-SMA、BAX蛋白表达均升高,COL1A1、BCL-2蛋白表达均降低,差异均有统计学意义(均为P<0.05);与缺氧12 h组相比,缺氧48 h组细胞COL1A1、BCL-2蛋白表达降低,BAX蛋白表达升高,差异均有统计学意义(均为P<0.05),其余蛋白表达变化不明显,差异均无统计学意义(均为P>0.05)(图1)。
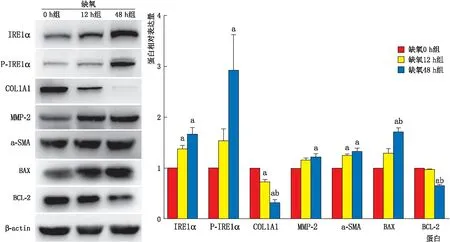
图1 Western blot检测各组细胞IRE1α、P-IRE1α、COL1A1、MMP-2、α-SMA、BAX及BCL-2蛋白表达情况 A:Western blot灰度图;B:各种蛋白相对表达量。与缺氧0 h组相比,aP<0.05;与缺氧12 h组相比,bP<0.05。
2.2 各组细胞α-SMA蛋白免疫荧光染色结果免疫荧光染色结果显示:缺氧12 h组及缺氧48 h组细胞α-SMA蛋白荧光强度均较缺氧0 h组增高,且缺氧48 h组较缺氧12 h组细胞α-SMA蛋白荧光强度升高更明显(图2)。
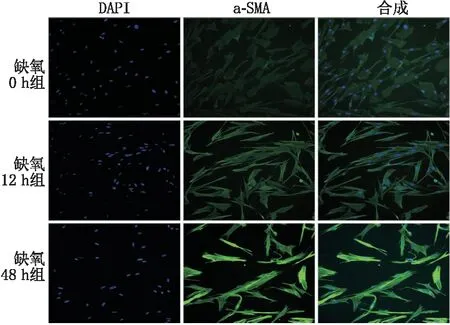
图2 各组细胞α-SMA蛋白免疫荧光染色结果(×200) 免疫荧光染色中α-SMA显示为绿色,DAPI 显示为蓝色。
2.3 各组细胞凋亡情况流式细胞仪检测结果显示:缺氧0 h组、缺氧12 h组、缺氧48 h组细胞凋亡率分别为(0.617±0.032)%、(2.187±0.212)%、(4.130±0.395)%;缺氧12 h组及缺氧48 h组细胞凋亡率均高于缺氧0 h组,差异均有统计学意义(均为P<0.05),且缺氧48 h组细胞凋亡率高于缺氧12 h组,差异有统计学意义(P<0.05)(图3)。
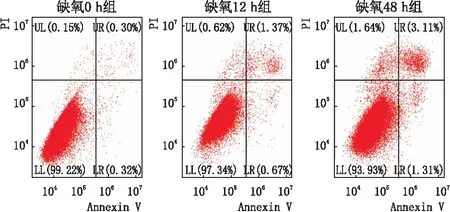
图3 流式细胞仪检测各组细胞凋亡情况
2.4 各组细胞增殖情况CCK-8检测结果显示:缺氧0 h组、缺氧12 h组、缺氧48 h组细胞D450分别为1.456±0.132、0.709±0.054、0.544±0.049;缺氧12 h组及缺氧48 h组细胞D450均低于缺氧0 h组,差异均有统计学意义(均为P<0.05);缺氧48 h组与缺氧12 h组相比,差异无统计学意义(P>0.05)(图4)。
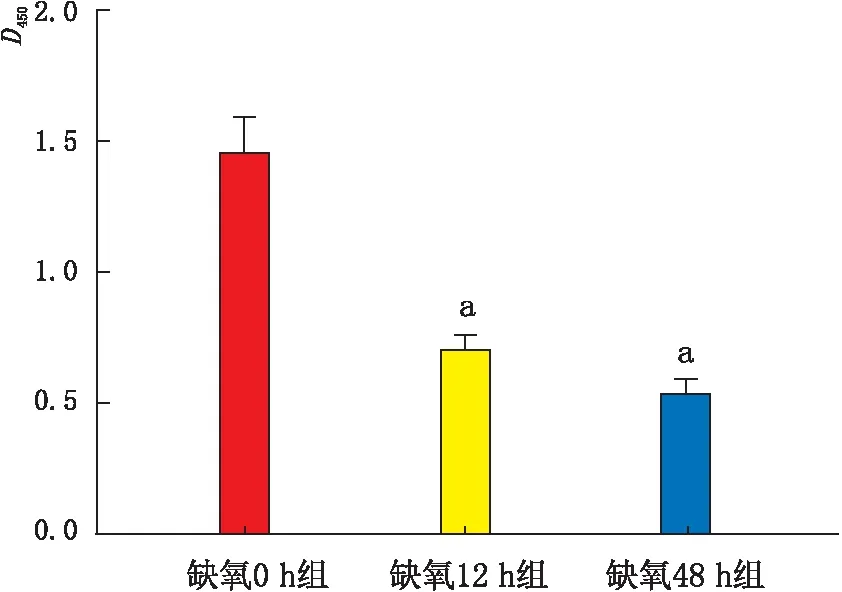
图4 CCK-8检测各组细胞增殖情况 与缺氧 0 h 组相比,aP<0.05。
3 讨论
近年来,大量研究表明,巩膜细胞外基质重塑在近视发生发展中起着至关重要的作用。巩膜细胞外基质主要由 I型胶原蛋白组成,巩膜重塑主要表现为 I 型胶原蛋白表达降低[4],参与这一过程的触发因素目前尚不清楚。最近研究发现,在近视发展过程中会出现脉络膜变薄、血流减少等现象,而脉络膜作为巩膜血液供应的重要来源其变薄可导致巩膜组织缺氧[5],并且采用单细胞测序分析技术也发现近视巩膜中缺氧信号富集,由此证明巩膜在近视发生发展中确实存在着缺氧现象[3]。巩膜成纤维细胞作为巩膜的主要细胞成分,可以合成、分泌各种细胞因子和大量细胞外基质,在维持巩膜形态中起重要作用[6]。因此,本研究利用缺氧处理HFSF以模拟近视眼巩膜缺氧环境,从细胞水平探讨缺氧对巩膜的影响及具体机制。我们的研究发现,缺氧激活体外培养的HFSF内质网应激反应,并可能通过调节HFSF胶原代谢、转分化及凋亡来参与巩膜重塑。
内质网是真核细胞中游离钙储存、蛋白质折叠和脂质合成的主要细胞器[7]。各种生理和病理刺激或损伤均可扰乱内质网稳态,致使错误折叠的蛋白在内质网累积,导致内质网应激的发生[8]。内质网应激主要表现为未折叠蛋白反应(UPR)[8]。UPR主要由3种跨膜蛋白,IRE1α、活化转录因子6和蛋白激酶RNA样内质网激酶调控[7],其中IRE1α是UPR的主要参与者[9]。生理情况下,IRE1α与内质网腔的伴侣蛋白GRP78结合而处于失活状态,当内质网应激发生时,GRP78与IRE1α解离,导致IRE1α自身磷酸化而激活下游信号蛋白,启动内质网应激相关的信号转导[10]。我们研究发现,缺氧导致IRE1α表达上调,同时其磷酸化蛋白P-IRE1α水平也随缺氧时间延长逐渐升高,证实激活了内质网应激反应。UPR主要通过抑制蛋白质的合成,增加蛋白质折叠能力和促进错误折叠蛋白的降解来恢复细胞内蛋白质稳态,从而维持细胞的正常生理功能,促进细胞存活[10]。目前研究表明,内质网应激可降低软骨细胞和真皮成纤维细胞胶原的产生[11],本实验中我们也证实了缺氧状态下巩膜成纤维细胞COL1A1蛋白表达下调,且缺氧时间越长,其下调越明显,这与之前的研究结果一致[12]。同时我们发现,缺氧导致 MMP-2表达上调,与既往研究发现形觉剥夺性近视小鼠模型中MMP-2表达明显升高一致[13]。MMP-2 可降解巩膜Ⅰ型胶原蛋白,导致胶原变薄、细胞外基质成分改变,致使巩膜生物力学强度下降,眼球易于扩张,从而导致近视的发生[13]。而已有研究证实,内质网应激可在转录和翻译水平上调MMP-2 的表达[14],再次证明内质网应激的确参与了缺氧诱导的巩膜重塑。此外,研究发现,近视发生时巩膜成纤维细胞转分化为肌成纤维细胞[15],为了进一步探讨缺氧在巩膜重塑中的作用,我们检测了成纤维细胞转分化标志物α-SMA蛋白的表达,发现缺氧状态下α-SMA表达升高,表明缺氧诱导了巩膜成纤维细胞向肌成纤维细胞转分化,而在肺纤维化及肾脏等疾病中已经证实内质网应激可促进成纤维细胞转分化[16-17]。基于上述结果我们推测缺氧诱导的内质网应激的确参与了近视的发生发展,但是具体的机制需要进一步验证。
UPR被认为是细胞对外界刺激的一种保护机制,但是当内质网应激过强或者持续存在时,UPR无法纠正失衡的蛋白质稳态,则会启动细胞凋亡相关的信号转导,并引发疾病[18]。长时间的内质网应激通过持续激活IRE1的磷酸激酶活性,致使JNK磷酸化激活,被磷酸化的JNK可抑制抗凋亡基因BCL-2的表达,同时促进促凋亡基因BAX的表达[18]。已有研究证实,内质网应激及其诱导的凋亡与心肌缺血缺氧再灌注损伤密切相关[19]。而我们的研究也证实,在缺氧条件下凋亡抑制蛋白BCL-2表达下调,同时促凋亡蛋白BAX表达明显上调,且随着缺氧时间的延长,细胞凋亡率逐渐增加,进一步证明缺氧激活了内质网应激反应并参与调节缺氧状态下巩膜成纤维细胞的存活。
总之,我们的研究结果表明,缺氧激活HFSF内质网应激反应,并且可能通过调节胶原代谢、促进细胞转分化及凋亡来参与巩膜重塑。本研究为近视患者巩膜重塑的机制提供了新的见解,并提示抑制内质网应激可能是控制病理性近视的一个潜在靶点。
猜你喜欢
医学研究生学报(2022年3期)2022-11-27
临床眼科杂志(2022年1期)2022-11-25
临床眼科杂志(2022年2期)2022-11-24
井冈山大学学报(自然科学版)(2022年1期)2022-02-28
眼科新进展(2021年12期)2022-01-15
医学研究杂志(2021年12期)2021-11-30
临床眼科杂志(2021年4期)2021-11-29
临床眼科杂志(2021年5期)2021-11-29
云南医药(2021年3期)2021-07-21
药学研究(2021年3期)2021-04-20
