
Neutrophil extracellular traps participate in the development of cancer-associated thrombosis in patients with gastric cancer
2022-07-30 10:03:08LiJCZouXMYangSFJinJQZhuLiCJYangZhangAGZhaoTQChenCY
World Journal of Gastroenterology 2022年26期
Li JC, Zou XM, Yang SF, Jin JQ, Zhu L, Li CJ, Yang H, Zhang AG, Zhao TQ, Chen CY
Abstract
Key Words: Neutrophil extracellular traps; Gastric cancer; Platelet; Endothelial cells; Venous thromboembolism
lNTRODUCTlON
Gastric cancer (GC) is one of the most prevalent gastrointestinal tumors and the third most fatal cancer in the world[1,2]. Additionally, venous thromboembolism (VTE) is a more common complication among GC patients when compared to healthy individuals[3-5]. Notably, VTE is associated with a high mortality in GC patients[6,7]. Several factors, such as tumor stage, neoadjuvant chemotherapy, and surgery, contribute to the development of VTE in GC patients[8,9]. Cancer cells exert a procoagulant activity (PCA) in their microenvironment, which is related to activation of the coagulation system[10].However, the molecular mechanism underlying PCA in GC patients is poorly understood. Uncovering molecular targets associated with VTE in GC patients can help in the development of appropriate therapy, which can improve the clinical outcomes of these patients.
Neutrophil extracellular traps (NETs) are web-like structures composed of filamentous DNA and histones, decorated with antimicrobial protein granules and enzymes, including neutrophil elastase(NE), myeloperoxidase (MPO), matrix metalloproteinase-9 (MMP-9), and cathepsin G (CatG)[11,12].They result from interactions between neutrophils and bacterial or other stimulating factors[13,14].Overall, they protect the host from pathogen-related damage. NETs were initially described as an antimicrobial reaction, but some undesirable effects of NETs have been reported in autoimmune diseases[15-17]. Citrullinated histone H3 (citH3) is proposed as a biomarker reflecting NETs formation.Recent studies have linked NETs to the development of metastasis and cancer-associated thrombosis[18,19]. In particular, NETs result in arterial and venous thrombosis, both of which are mediated by neutrophils[20]. Furthermore, using mouse models, it has been shown that thrombosis in breast cancer tissues is closely linked to the formation of NETs[21]. This finding suggests the potential relationship between NETs and cancer-associated thrombosis.
A recent study revealed that priming metastatic pancreatic cancer cells with platelets stimulates neutrophils to release NETs, which promotes thrombosis in both static and dynamic states[22]. We first reported that platelets derived from GC can stimulate neutrophils to release NETs. NETs enhance PCA in GC patients, which is positively correlated with the expression of the thrombin-antithrombin (TAT)complex and the level of serum D-dimers[23]. Moreover, both human and animal studies suggest that enhanced thrombosis may result from increased activated platelets[24,25]. Nevertheless, little is known about the interaction between NETs and platelet activation in GC patients. Venous endothelial cell (EC)injury in cancer patients is also closely related to venous thrombosis[26,27]. Interestingly, the cytotoxicity of NETs against ECs enhances PCA in oral squamous cancer, even in patients with obstructive jaundice and inflammatory bowel disease[28-30]. Even so, the potential mechanism underlying EC injury in GC patients is poorly understood.
Our central hypothesis is that GC-induced NETs participate in VTE responses by platelet activation and endothelial injury. Therefore, we first explored the complex relationship between NET formation and platelet activation as well as EC injury. Furthermore, we showed the effect of NETs on thrombosis in an inferior vena cava (IVC) stenosis mouse model. In general, our results may indicate that NETs are potential therapeutic targets in the prevention and treatment of VTE in GC patients.
MATERlALS AND METHODS
Patients and tissue samples
Sixty-three patients newly diagnosed with primary GC and 13 healthy donors (HDs) attending the Second Affiliated Hospital of Harbin Medical University between October 2019 and April 2021 were enrolled in this study. GC diagnoses were performed based on pathological examinations. Pathological tumor-node-metastasis (TNM) staging and histological classification of GC were performed according to the 7thAmerican Joint Committee on Cancer (AJCC) guidelines[31]. Patients who were < 18 years old or pregnant, those on antitumor or anticoagulant treatment before surgical treatment, or those with underlying complications such as endocrine, cardiovascular, hematological and other cancers or infectious disease were all excluded. The main clinical characteristics of the patients with GC and HDs are shown in Table 1. We extracted tumor and adjacent normal tissues from GC patients who consented to this study in writing. Blood samples of preoperative patients were obtained at first diagnosis before any clinical treatment, and blood samples of postoperative patients were obtained from 1 mo after surgery before adjuvant therapy to avoid errors caused by postoperative stress. The protocol for this study was approved by the Ethics Committee of the Second Affiliated Hospital of Harbin Medical University (No. KY2016-032).
Isolation of human plasma, platelets, and neutrophils
Fresh whole venous blood was collected into tubes containing 3.2% sodium citrate using 21-gauge needles. The patients underwent overnight fasting before blood collection. The blood was centrifuged at 150gfor 20 min at room temperature (RT) to obtain platelet-rich plasma (PRP), within 1 h of collection.Clean top PRP layer was collected in a new tube, diluted with platelet wash buffer (TBD, Tianjin,China), and centrifuged at 460gfor 20 min at RT to obtain clean platelets and platelet-free plasma (PFP)[32]. The platelets were resuspended in prewarmed modified Tyrode’s buffer (137 mmoL/L NaCl, 2.8 mmoL/L KCl, 1.0 mmoL/L MgCl2, 12 mmoL/L NaHCO3, 0.4 mmoL/L Na2HPO4, 5.5 mmoL/L glucose,pH 7.4; Solarbio, Beijing, China). Neutrophils were isolated based on the density gradient centrifugation, using the whole blood neutrophil isolation kit (TBD). After lysing erythrocytes based on the red blood cell lysis buffer (150 mmoL/L NH4CL, 10 mmoL/L KHCO3, 0.2 mmoL/L Na2EDTA, pH 7.4;TBD), the purity (> 96%) and viability (> 96%) of neutrophils were assessed using Wright-Giemsa staining and Trypan blue staining, respectively.
Cell lines and conditioned medium
The human metastatic GC KATO-III and MKN-45 cell lines, human primary GC AGS cell line, human gastric mucosal epithelial cell (GES-1) line, human umbilical cord endothelial cell (HUVEC) line, and mouse forestomach squamous carcinoma (MFC) cell line were purchased from PROCELL (Wuhan,China). All cell lines were characterized using short tandem repeat (STR) profiling. The GES-1, AGS,MKN-45 and MFC cells were cultured in RPMI 1640 (Gibco, USA), KATO-III cells were cultured in IMDM (Gibco), and HUVECs were cultured in DME/F12 (HyClone, Logan, UT, USA). All media were supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin-streptomycin solution(Beyotime, Beijing, China). Incubation was performed at 37°C under 5% CO2in a humidified environment. To prepare conditioned medium (CM), the cells were cultured to 90% confluence in medium supplemented with 10% FBS, washed three times using 1 × PBS, and recultured for 48 h in medium without FBS. The supernatant was centrifuged at 1500gfor 10 min at 4°C to remove cell debris.The CM was collected and stored at -80°C until use.
Animal models
The animal protocol was designed to minimize pain or discomfort to the animals. Wild-type male C57BL/6 mice (7-9 wk old, weighing 20-26 g) were purchased from the Animal Experimental Center of Harbin Medical University, and all procedures were approved by the Animal Care and Use Committeeof the Second Affiliated Hospital of Harbin Medical University (No. KY2016-032). The animals were individually housed and maintained under standard conditions (12 h/12 h light/dark cycle, 22 ± 1 °C,50% humidity) and provided with a conventional laboratory diet and an unrestricted supply of drinking water. In the subcutaneous tumor models, mice were injected with MFC cells (2 × 107cells/mL)subcutaneously into the right axilla, and the tumor volumes were measured every 3 d from 7 d after injection with MFC cells and allowed to reach 1000 mm3(usually 28-35 d). The volume was calculated by measuring the length (L) and width (W) of the tumor: Tumor volume = π/6 × L × W2. Thereafter, a murine model of deep vein thrombosis (DVT) was developed as previously described[33]. The mice were anesthetized by intraperitoneal injection of 2,2,2-tribromoethanol (Sigma, St . Louis, MO, USA),and the intestines were removed to expose the IVC after entering the abdominal cavity through a median abdominal incision. The IVC was carefully separated below the left renal vein plane. After 5-0 (1 mm) sutures passed through the IVC, 3-0 (2 mm) sutures were placed at the parallel part of the IVC as the blocking line. The IVC was ligated and 3-0 sutures were carefully extracted. This procedure has been shown to decrease the vascular lumen by approximately 90%. The other branches of the IVC were ligated to the level of the iliac vein. Thereafter, the abdominal incision was closed, mice were sacrificed after 6 or 48 h, and thrombi that had formed in the IVC were harvested. The IVC stenosis mice in the treatment groups were injected with DNase I (50 μg/mouse; Roche, Switzerland) intraperitoneally every 12 h until the time of death. All animals were killed for tissue collection. Blood (300 μL/mouse)was drawn from the periorbital eye plexus and stabilized with 0.5 mmoL/L EDTA. Plasma was obtained as described above. Thrombi of control and tumor-bearing mice were fixed in 4% paraformaldehyde for 24 h, embedded in Optimal Cutting Temperature (OCT) compound (SAKURA, Torrance,CA, USA), and cryosectioned at a thickness of 4 μm for immunofluorescence staining.
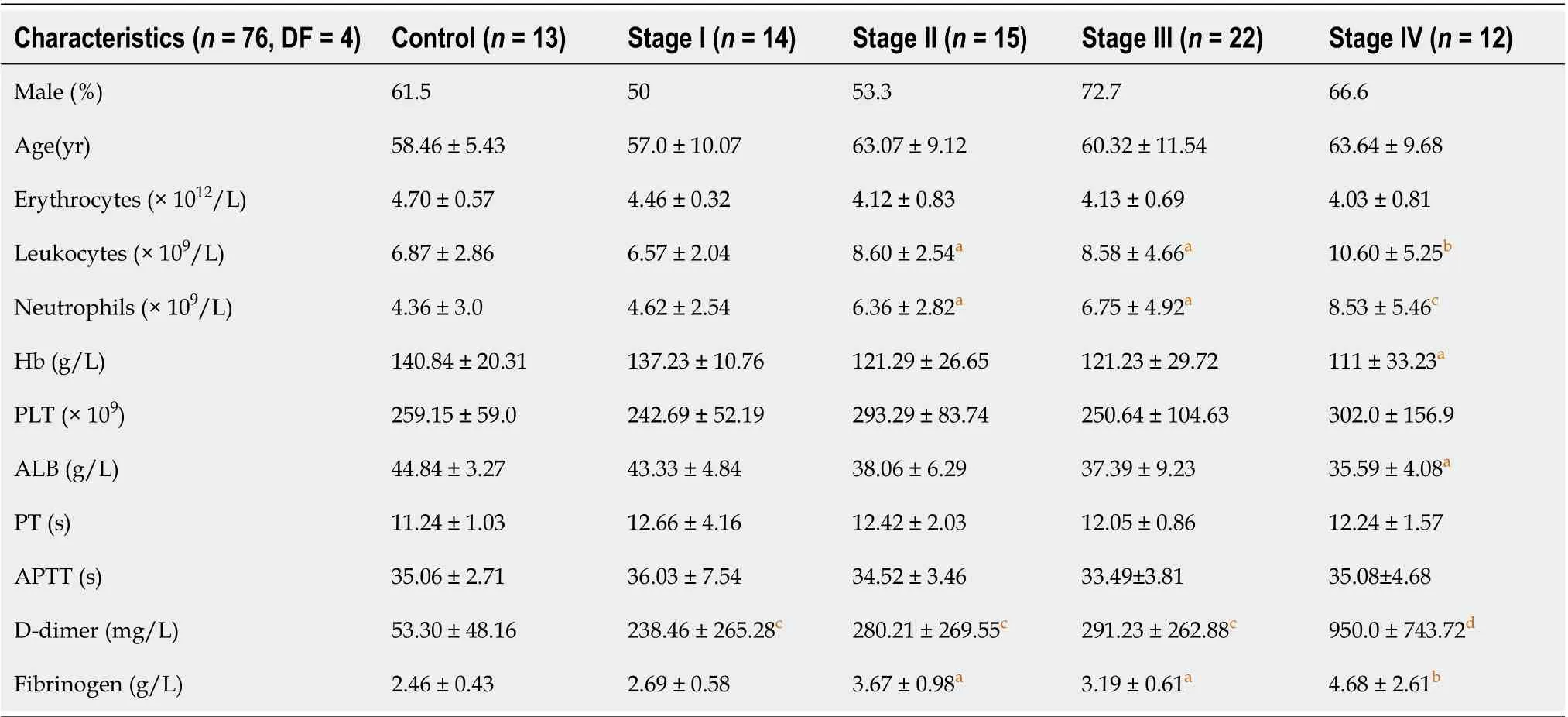
Table 1 Main clinical and laboratory features of 13 healthy subjects and 63 patients diagnosed with gastric cancer
Quantification of plasma NETs marker
Plasma cell-free DNA (cf-DNA), MPO-DNA and citH3-DNA complexes were quantified using capture ELISA as previously described[34]. The quantification of cf-DNA was performed using the Quant-iT PicoGreen dsDNA assay kit (Invitrogen, Carlsbad, CA, USA). For detection of NET-DNA complexes, 5 μg/mL anti-MPO (ab90810; Abcam, Cambridge, UK) or anti-citH3 antibody (ab5103; Abcam) was coated onto 96-well plates overnight at 4°C. After blocking in 1% bovine serum albumin (BSA), the plasma from GC patients or healthy individuals was added per well and incubated at RT for 2 h. After washing five times with PBST, Quant-iT PicoGreen dsDNA Reagent was added. The values were then read with a fluorometer with a filter setting of 480 nm/520 nm excitation/emission wavelengths.
Fibrin formation and TAT complex assay
Fibrin formation of platelets and ECs was detected by turbidity as previously described[35]. To assess the fibrin formation in the setting of platelet or EC monolayers, cell monolayers in 96-well plates were stimulated by NETs with or without DNase I, activated protein C (APC), and sivelestat treatment alone or together for 4 h, followed by two washes with Hank’s balanced salt solution (137 mmoL/L NaCl, 5.3 mmoL/L KCl, 4.17 mmoL/L NaHCO3, 0.33 mmoL/L Na2HPO4, 5.5 mmoL/L glucose, pH 7.4) and then 150 μL PFP from HDs was cocultured with cells at 37°C for 2 min, followed by the addition of 50 μL prewarmed 25 mmoL/L CaCl2. The fibrin formation was tested using turbidity measurements and quantified as the maximum value by measuring the OD at 405 nm every 10 s for 30 min on a SpectraMax 340 PC plate reader. To assess the fibrin formation in the plasma of mice, 150 μL PFP from control, tumor-bearing mice, or DNase I infused tumor-bearing mice was cultured at 37°C for 2 min,followed by addition of 50 μL prewarmed 25 mmoL/L CaCl2. To detect the level of TAT complex, a human and mouse TAT complex ELISA kit (Jingkbio, Shanghai, China) was used as previously described[36].
Flow cytometry
Circulating NETs were measured using flow cytometry. Here, whole blood from HDs and GC patients was diluted with 1 × PBS and incubated in the dark at RT for 30 min with FITC-conjugated-citH3(eBioscience, San Diego, CA, USA) and PE-conjugated MPO (eBioscience) antibodies. For the assessment of phosphatidylserine (PS) and P-selectin expression on platelets, platelets (2 × 106cells) isolated from the blood of HDs and GC patients were incubated with FITC-conjugated lactadherin (Haematologic,Essex Junction, VT, USA), APC-conjugated CD62P (Biolegend, San Diego, CA, USA), and PerCPconjugated CD41 (Biolegend) antibodies.
Immunofluorescence staining of NETs
Tumor and paratumor tissues of GC patients were fixed in 4% paraformaldehyde for 24 h, embedded in OCT compound (SAKURA), and cryosectioned into slices of 4-μm thickness. The samples were cultured overnight at 4°C with primary rabbit anti-histone H3 (1:500, ab5103; Abcam) and mouse anti-MPO(1:500, ab90810; Abcam) antibodies, and washed three times with PBS before incubation for 1 h at RT with Alexa Fluor 594-conjugated goat anti-rabbit (1:200; Proteintech, China) and Alexa Fluor 488-conjugated goat anti-mouse (1:200; Proteintech) secondary antibodies. The tissues were stained for 5 min at RT in the dark with 4’,6-diamidino-2-phenylindole (DAPI) and anti-fade mounting medium(Solarbio, Beijing, China). Thrombi in the IVC of tumor models or control mice were stained with primary rabbit anti-histone H3 (1:500, ab5103; Abcam) and rat anti-Ly6G (1:500, Novus, St. Charles, MO,USA), and the specimens were incubated with the Alexa Fluor 488-conjugated goat anti-rabbit (1:200;Proteintech) and Alexa Fluor 594-conjugated goat anti-rat (1:200; Proteintech) secondary antibodies as previously described[37]. The tissue images were captured using a confocal microscope (LSM 800; Zeiss,Germany).
Neutrophils (5 × 105cells) isolated from HDs were seeded and incubated in glass-based poly-L-lysinecoated 24-well plates for 1 h at 37°C under 5% CO2. Thereafter, cell suspensions of KATO-Ⅲ, MKN-45,AGS, and GES-1 (2 × 105cells) or CM from GC cells were cocultured with neutrophils for 4 h at 37°C under 5% CO2. To detect and quantify NETs, the samples of the CM group were incubated with primary rabbit anti-histone H3 and mouse anti-MPO antibodies and then fluorescent secondary antibodies. For the samples of cell-cell contact groups, NETs were stained with Sytox Green (Solarbio) for 10 min in the dark at RT. All images were captured using a confocal microscope (LSM 800; Zeiss) and analyzed with ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Preparation of cell-free NETs
Cell-free NETs were isolated from neutrophils of GC patients as previously described, with modifications[38]. Neutrophils (107cells/mL) were cultured for 4 h at 37°C under 5% CO2in medium supplemented with 500 nM PMA (HY-18739; MedChemExpress, Monmouth Junction, NJ, USA). The supernatant was discarded, and ice-cold 1 × PBS was added to wash down the cell layer of neutrophils to obtain the NET medium and centrifuged at 1500gfor 10 min at 4°C to remove cell debris. Thereafter,1.5 mL supernatant (sterile DNA-protein complex) was centrifuged at 15 000gfor 15 min at 4°C. The resultant pellets were suspended in ice-cold 1 × PBS, followed by DNA concentration measurement in the medium obtained using spectrophotometry (Biospec-nano, Japan). An adequate DNA concentration in the medium should range between 50 and 100 μg/mL. The medium containing the NETs was stored at -80°C for subsequent experiments.
传统的对车辆综合传动装置的动力学研究,针对的主要是子系统或者零部件的动力学模型的建立和求解,不涉及虚拟装配的内容,更不具备实时性和交互性等特点。基于虚拟装配的实时动力学仿真,除了考虑逼真的场景模型,还需要对综合传动装置进行准确的动力学建模,该模型需要包含如零件几何信息、基本物理属性信息等多种信息,以满足动力学模型的准确性,从而能够实现综合传动装置在实时动力学仿真平台下进行实时动力学仿真。
Platelet activation and adhesion assays
Platelet activation and adhesion assays were performed as previously described[22]. Glass-based wells of 24-well plates were coated with cell-free NETs after overnight incubation in the corresponding medium at 4°C in a humidified chamber. For controls, 1% denatured BSA was used. Denatured BSA was prepared by heating the solution in PBS without calcium or magnesium to 80°C for 3 min and immediately placing it on ice until cool; it was then stored at 20°C until use. Platelet suspensions (107cells/mL) were then seeded in the wells, cultured for 1 h at 37°C under 5% CO2, fixed for 15 min at RT with 4% paraformaldehyde, and washed three times using 1 × PBS before 20 min permeabilization using 0.1% Triton-X 100. The platelets were then incubated for 30 min with Alexa Fluor 594-conjugated phalloidin primary antibody (1:300; Thermo Fisher, Waltham, MA, USA). To assess PS and P-selectin expression, the platelet suspension was incubated for 1 h at 37°C under 5% CO2, and the cells were stimulated with NETs for 1 h, followed by FITC-conjugated lactadherin (Hematologic) staining for 30 min. Platelets were washed twice with PBS before fixation for 15 min at RT with 4% paraformaldehyde,permeabilized using 0.1% Triton-X 100 for 20 min, and then stained with primary rabbit anti-P-selectin(1:200; Proteintech) and mouse anti-CD41 (1:500; Novus) antibodies. Images were captured using a confocal microscope (LSM 800; Zeiss) and analyzed with ImageJ software (National Institutes of Health).
HUVECs stimulation assay
HUVECs were incubated with cell-free NETs or PBS in 24-well plates for 4 h when the cells grew to a confluent monolayer. The cells were fixed in 4% paraformaldehyde for 15 min at RT, washed three times using 1 × PBS, and blocked for 1 h using 10% goat serum with 1% BSA solution in PBS. For detection of tissue factor (TF) expression, ECs were incubated overnight at 4 °C with rabbit anti-TF (1:500, ab228968;Abcam) and mouse anti-CD31 (1:500, ab9498; Abcam) primary antibodies. The cells were washed with PBS and reincubated for 1 h at RT with Alexa Fluor 594-conjugated (Proteintech) goat anti-rabbit and Alexa Fluor 488-conjugated (Proteintech) goat anti-mouse secondary antibodies. For detection of intercellular junctions of cells, cells were incubated overnight at 4°C with rabbit anti-VE-cadherin (1:500,ab33168; Abcam) primary antibody, followed by Alexa Fluor 488-conjugated (Proteintech) goat antirabbit secondary antibody, and were further incubated with Alexa Fluor 594-conjugated phalloidin primary antibody (1:300; Thermo Fisher, Waltham, MA, USA). They were stained with DAPI and fixed with mounting medium (Solarbio) for 5 min at RT in the dark. The cells were observed and photographed using a confocal microscope. The photos were analyzed with ImageJ software.
Platelet and HUVEC inhibition assays
For inhibition assays, platelets and HUVECs were cocultured with cell-free NETs for 1 h at 37°C in a humidifier chamber in the presence of DNase I (100 U/mL, Roche), APC (100 nM, HY-P1918;MedChemExpress), and sivelestat (100 nM, HY-17443; MedChemExpress) alone or together. DNase I cleaves NET DNA, whereas APC and sivelestat disrupt histones and NE functions, respectively.
Statistical analysis
Data are expressed as the mean ± SD. Data were analyzed to assess distribution normality. For normally distributed data, statistical significance was analyzed using Student’st-tests and one-way analysis of variance. For non-normally distributed data, statistical significance was analyzed using the Mann-Whitney test and Kruskal-Wallis test. All analyses were performed using GraphPad Prism version 8.0 and SPSS 16.0 statistical software. Spearman’s correlation was used to evaluate the association between two variables.P< 0.05 was considered statistically significant.
RESULTS
GC patients display greater NET formation
The levels of plasma cf-DNA, citH3-DNA,and MPO-DNA complexes in GC patients (n= 63) and HDs (n= 13), which reflect the concentration of NETs, were measured using capture ELISA. The levels of NET markers were significantly higher in patients with stage II/III/IV GC than in HDs (Figure 1A-C). There was also a significant difference in preoperative and postoperative plasma NET marker levels in GC patients (Figure 1D-F). The levels of NET markers positively correlated with those of serum D-dimer(cf-DNA:r= 0.5595,P< 0.0001; citH3-DNA:r= 0.5469,P< 0.0001; MPO-DNA:r= 0.5479,P< 0.0001),suggesting that NETs were associated with hypercoagulation and VTE development in GC patients. The levels of NETs (MPO+/citH3+neutrophils) in the circulation in GC patients and HDs were measured using flow cytometry. Circulating NETs were higher in the blood of patients with either GC stage(II-IV) than in their HD counterparts (Figure 1G and H). Furthermore, based on MPO and citH3 levels,immunofluorescence staining revealed that NETs were significantly higher in the tumor microenvironment than in the paratumor tissue of the same patients (Figure 1I-M).
GC cells stimulate formation of NETs by neutrophils
To assess whether GC cells directly stimulated NET formation, we analyzed the expression of NETs in a coculture of GC cell lines and neutrophils. Immunofluorescence analysis revealed that compared with GES-1 cells, the rate of NET formation was significantly higher in GC cells (Figure 2A). However, the formation of NETs was greater in the metastatic GC cell line than in the nonmetastatic GC cell line(Figure 2B). We measured NET formation when normal neutrophils were cocultured with CM from KATO-III, MKN-45, AGS and GES-1 cells. Immunofluorescence analysis further revealed that the CM of KATO-III and MKN-45 cells exerted greater neutrophil activation for NET formation than the CM of AGS cells. However, the CM of GES-1 cells had no effect on NET formation (Figure 2C and D). Overall,these findings demonstrate that GC cells stimulate NET formation through both intercellular contact and noncontact mechanisms.
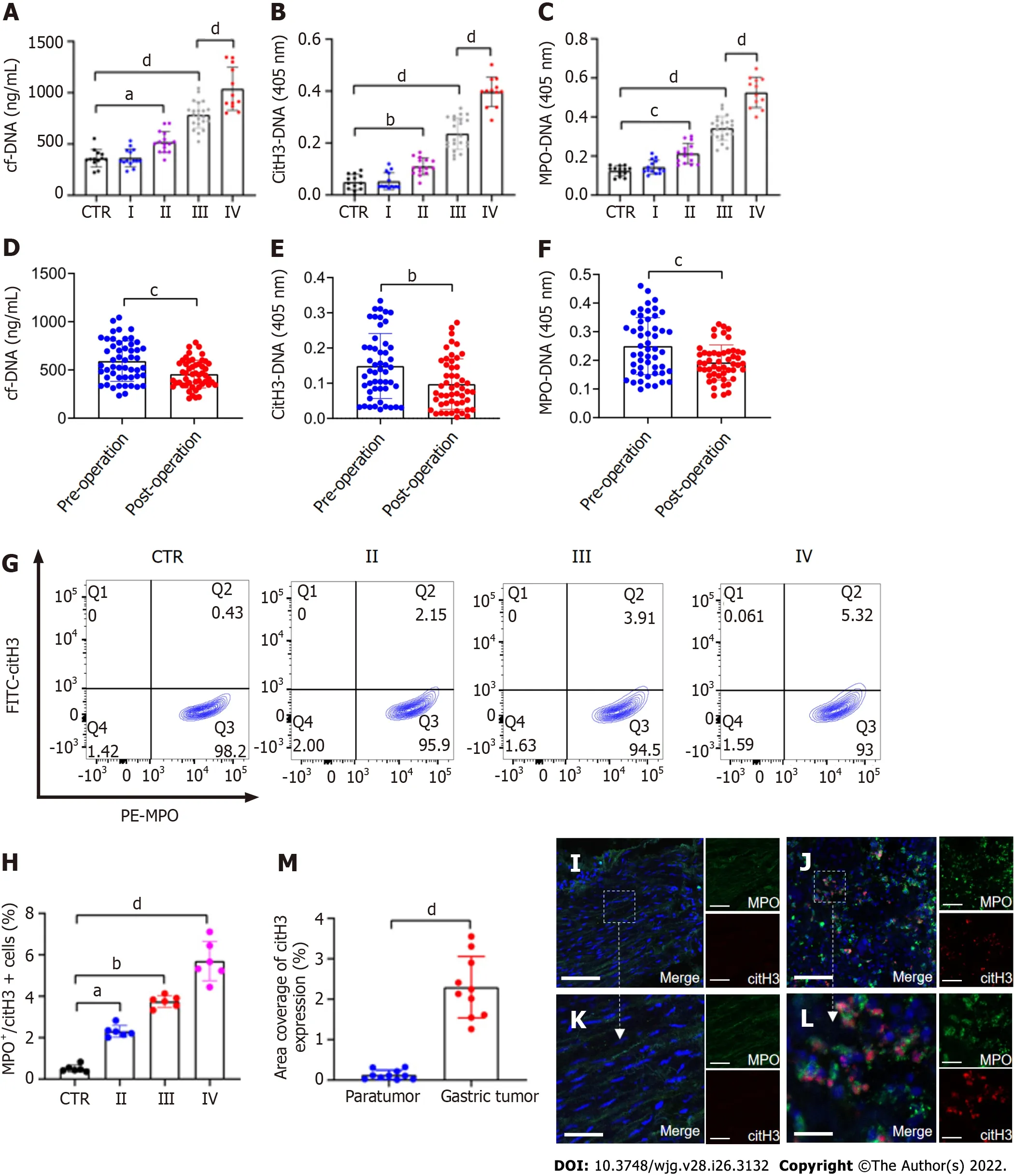
Figure 1 NETs are accumulated in samples of GC patients. A-C: Plasma levels of NET markers cf-DNA, citH3-DNA, and MPO-DNA in GC patients and healthy individuals were measured by ELISA. Healthy individuals, n = 13; stage I, n = 14; II, n = 15; III, n = 22; IV, n = 12; D-F: Comparison of cf-DNA, citH3-DNA and MPO-DNA in the plasma of patients with GC preoperatively and postoperatively by ELISA. n = 51; G and H: The rate of activated neutrophils in the circulating environment of GC patients and healthy individuals was measured by flow cytometry with APC-MPO and FITC-citH3 staining. Each group: n = 6; I and J: NET accumulation was detected by confocal microscopy with MPO and citH3 staining in paratumor and tumor samples from the same GC patient. Magnification 20×; scale bars: 50 μm. Red-citH3, Green-MPO and Blue-DAPI; K and L: Magnified (40×) part of MPO and citH3 colocation in paratumor and tumor samples from the same patient. Scale bars: 10 μm. Red-citH3, Green-MPO and Blue-DAPI; M: The percentage of area coverage of citH3 expression was defined as the rate of red area in total area and analyzed with ImageJ software; all values are the mean ± SD. aP < 0.05; bP < 0.01; cP < 0.001; dP < 0.0001. NET: Neutrophil extracellular trap; GC:Gastric cancer; cf-DNA: Cell-free DNA; MPO: Myeloperoxidase; citH3: Citrullinated histone H3.
NETs contribute to hypercoagulation of platelets

Figure 2 GC cells can stimulate neutrophils to form NETs. A and B: Control neutrophils were cocultured with normal gastric mucosal epithelial cells (GES-1) or GC cells (AGS, MKN-45 and KATO-III) and NET formation was measured by confocal microscopy with cell-impermeable Sytox-Green staining. Magnification 20×; scale bars: 50 μm. Green: Neutrophils; C and D: Control neutrophils were cocultured with CM from GES-1 or GC cells, and stained with MPO and citH3. The percentage of NET-releasing cells was defined as the ratio of the calculated NET releasing neutrophils to the total number of neutrophils. Magnification 20×; scale bars: 50 μm. Red-citH3, Green-MPO, and Blue-DAPI. All values are the mean ± SD. aP < 0.05; bP < 0.01; cP < 0.001; dP < 0.0001. GC: Gastric cancer; GES-1:Gastric mucosal epithelial cells; NET: Neutrophil extracellular trap; CM: Conditioned medium; MPO: Myeloperoxidase; citH3: Citrullinated histone H3.
To examine the effect of NETs on platelet activation, we measured the levels of PS and P-selectin expression in these cells. Flow cytometry revealed that compared to HDs, the expression of PS and Pselectin was significantly higher on platelets of patients with GC (Figure 3). In addition, platelets isolated from HDs were cocultured with NET medium or PBS before analyzing PS and P-selectin expression. We found that NETs stimulated PS and P-selectin expression on platelets by confocal microscopy (Figure 4A-C). Flow cytometry also demonstrated this hypercoagulable phenotype of platelets which was stimulated by NETs (Figure 4D and E). In the inhibition assay, we added DNase I,APC and sivelestat alone or together to cleave DNA, histones and NE, which were the most functional factors of NETs. At the highest concentration of NETs (0.5 μg DNA/mL), flow cytometry revealed that DNase I, APC, sivelestat, or a combination of the three inhibited 60.2%, 47.1%, 41.9% and 83.2% of PS expression, respectively (Figure 4D), and 55.2%, 47.0%, 41.0%, and 91.76% of P-selectin expression on platelets, respectively (Figure 4E).
NETs promote platelet adhesion and prothrombotic state
Previous studies have shown that NETs promote thrombosis in murine late-stage breast cancer models and DVT models[19,39]. However, whether NETs derived from GC neutrophils have the ability to stimulate platelet adhesion under static conditions is unknown. To determine the effect of NETs on platelet adhesion, platelets isolated from HDs were seeded in NET-coated wells to measure the effects of NETs on the adherence of platelets to blood vessels. Confocal microscopy revealed that NETs enhanced adherence of platelets to glass slides (Figure 5A and B), indicating that NETs induce the development of thrombosis. Moreover, the results revealed that fibrin formation and TAT complex levels were increased when control plasma was cocultured with platelets activated by NETs (Figure 5C and D).
Inhibition assays revealed that digestion of NET DNA using DNase I modulated the adhesion of platelets on glass surfaces. Even so, a few platelets still adhered to the NET-coated well pretreated with DNase I (Figure 5A). This suggested that other protein components other than NET-DNA participated in the adhesion of platelets. NETs treated with DNase I, APC, sivelestat, or a combination of the three inhibited 67.1%, 56.6%, 38.9%, and 91.8% of platelet adhesion, respectively (Figure 5B). The degree of platelet adhesion in the combination group was comparable to that of the controls. We found that DNase I, APC, sivelestat, or a combination of the three reduced fibrin formation by 81.1%, 73.9%, 64.3%,and 90.7%, respectively (Figure 5C), and inhibited 78.9%, 57.4%, 51.2%, and 91.9% of TAT complex level,respectively (Figure 5D), at the highest concentration of NETs. Taken together, these findings demonstrate that NETs play a role in the development of thrombosis.
NETs drive hypercoagulation of ECs

Figure 3 Platelets are activated in patients with GC. A and C: Flow cytometry showed the rate of P-selectin-positive platelets from each stage GC patients and healthy individuals; B and D: Rate of phosphatidylserine-positive platelets from each stage GC patients and healthy individuals. All values are the mean ± SD. dP< 0.0001. PS: Phosphatidylserine; GC: Gastric cancer. CD62P: P-selectin; Lact: Lactadherin.
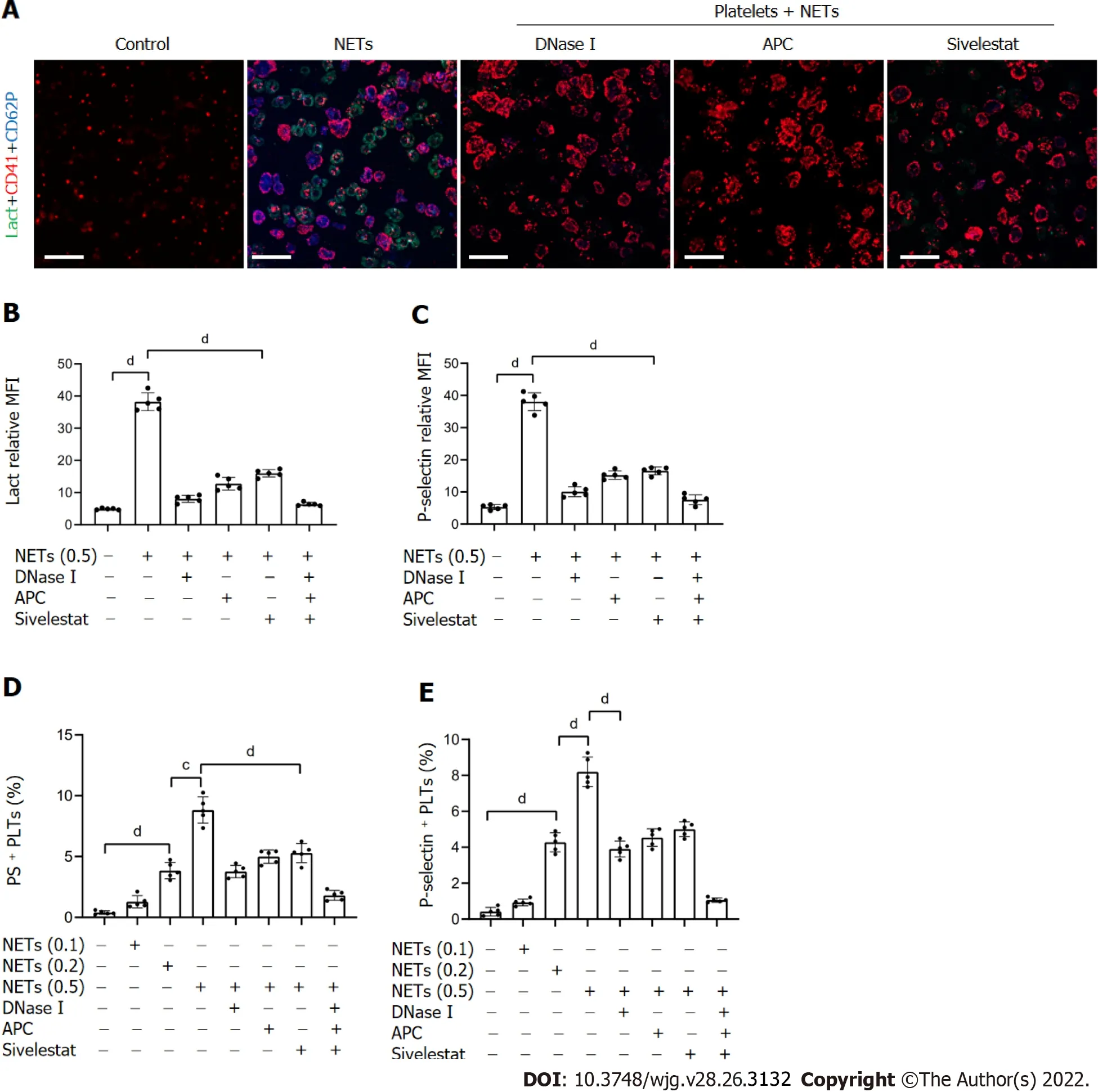
Figure 4 NETs contribute to hypercoagulation of platelets. A: PS exposure and P-selectin expression were measured when isolated platelets were cocultured with BETs (μg, DNA/mL) or in the presence of DNase I, activated protein C, and sivelestat alone or together by confocal microscopy. Magnification 63×;scale bars: 10 μm. Red-platelets, Green-Lactadherin, and Blue-P-selectin; B and C: PS exposure and P-selectin expression are indicated as MFI. MFI was defined as the ratio of total fluorescence intensity to the area; D and E: The rates of PS-positive platelets and P-selectin-positive platelets were detected by flow cytometry. All values are the mean ± SD. cP < 0.001; dP < 0.0001. PS: Phosphatidylserine; GC: Gastric cancer; DNase I: Deoxyribonuclease I; APC: Activated protein C; MFI: Mean fluorescence intensity; Lact: Lactadherin; CD41: Platelet; NET: Neutrophil extracellular trap.
To detect the effect of NETs on EC thrombogenicity, HUVECs were cocultured with NET medium.Confocal microscopy revealed that NETs destroyed the normal intercellular junctions between ECs(Figure 6A and C). NET treatment upregulated expression of TF on the surface membrane of ECs(Figure 6B and D). In addition, plasma fibrin formation and TAT complex levels were significantly increased when control plasma was incubated with EC monolayers activated by NETs (Figure 6E and F). Further inhibition assays were performed to assess the effect of NETs on ECs after pretreatment with DNase I, APC, sivelestat, or a combination of the three. We found that NET treatment after incubation with DNase I, APC, sivelestat, or all three drugs returned 47.5%, 36.3%, 33.4%, and 86.5%, respectively,of VE expression on ECs (Figure 6C), whereas similar treatment inhibited 60.4%, 44.8%, 38.6%, and 95.5%, respectively, of TF expression (Figure 6D). Additionally, we found that NET treat-ment after incubation with the above inhibitors inhibited 59.7%, 54.4%, 54.0%, and 91.5% of fibrin formation level,respectively (Figure 6E), and inhibited 51.2%, 35.6%, 25.9%, and 84.3% of TAT complex level,respectively (Figure 6F). Taken together, these findings suggest that NETs promote hypercoagulation of ECs; thus, inhibiting NET function can protect against venous injury.
NETs promote formation of thrombi in tumor-bearing IVC flow restriction mice
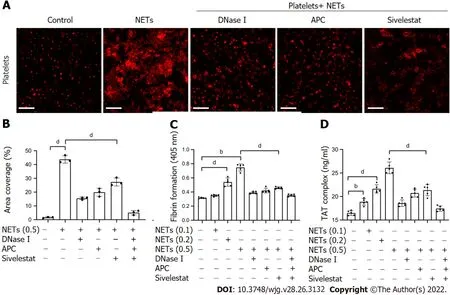
Figure 5 NETs promote platelet adhesion and prothrombotic state. A: Isolated platelets were incubated on glass slides which were coated with 1%dBSA, NETs (μg DNA/mL), or NETs pretreated with DNase I, APC, and sivelestat alone or together, followed by the F-actin components of platelets with 594-phalloidin staining. Magnification 63×; scale bars: 10 μm. Red-platelets; B: The percentage of area coverage of platelet adhesion was defined as the rate of red area in the total area and analyzed with ImageJ software; C: Isolated platelets were cocultured with different concentrations of NETs for 30 min with or without DNase I,APC and sivelestat treatment alone or together, and plasma fibrin formation was tested using turbidity measurements and monitored OD at 405 nm; D: TAT complex level of activated platelets was analyzed by ELISA. All values are the mean ± SD. aP < 0.05; bP < 0.01; cP < 0.001; dP < 0.0001. dBSA: Denatured bovine serum albumin; NETs: Neutrophil extracellular traps; DNase I: Deoxyribonuclease I; APC: Activated protein C; TAT: Thrombin-antithrombin.
Based on these findingsin vitroand the pivotal role of NETs in thrombosis, we hypothesized that GCinduced NETs can also promote thrombosisin vivo. Here, in a mouse IVC flow stenosis model, tumorbearing mice demonstrated more capacity to form thrombi and showed heavier weight and longer length of thrombi compared to control mice (Figure 7A-D). In the 6 h models, three of nine of control mice showed thrombi, whereas seven of nine of tumor-bearing mice formed thrombi. In the 48 h models, all mice demonstrated thrombi in the IVC. In addition, confocal images of thrombi formed in the tumor-bearing mice after 48 h of IVC stenosis showed that NETs were significantly accumulated compared to control mice (Figure 7E-H). The thrombi of control mice included some neutrophils(Ly6G+) but were not activated to form NETs (Figure 7E and G). Furthermore, tumor-bearing mice showed higher fibrin formation and TAT complex levels than control mice (Figure 7I and J).
In the inhibition assay, we infused DNase I into mice immediately after IVC stenosis and examined thrombosis after 6 or 48 h of surgery. We found that treatment with DNase I significantly inhibited thrombi formation in tumor-bearing IVC stenosis mice (Figure 7A-D). Furthermore, fibrin formation and TAT complex levels in tumor-bearing mice were significantly decreased by DNase I treatment(Figure 7I and J). These data suggest that NETs play a role in thrombosisin vivo, which was induced by GC, and inhibiting NETs by DNase I had a protective effect on thrombosis in this mouse model.
DlSCUSSlON
Inflammation is one of the hallmarks of cancer. Additionally, neutrophils are among the most important immune cells implicated in promoting tumor progression[40,41]. NETs participate in cancer progression by promoting the proliferation, invasion, metastasis and angiogenesis of cancer cells as well as thrombosis in numerous tumor types[42-44]. A recent study using mouse models with Jak2V617Fknock-in revealed that most myeloproliferative neoplasms display NET formation and DVT[45]. Our previous studies revealed that NETs promote the migration and metastasis of GC cells bothin vitroandin vivothrough epithelial mesenchymal transition[46]. Intriguingly, inhibition of NETs promotes apoptosis and inhibits the invasion of GC cells by regulating the expression of Bcl-2, Bax and nuclear factor-κB proteins[47]. Our initial studies revealed that NETs released by neutrophils in GC patients promoted the conversion of thrombin and fibrin[23]. Accordingly, we hypothesized that NETs promote thrombosis in GC patients. We investigated the interactions between GC cells, neutrophils, platelets and ECs, with a keen focus on their role in cancer-associated thrombosis.
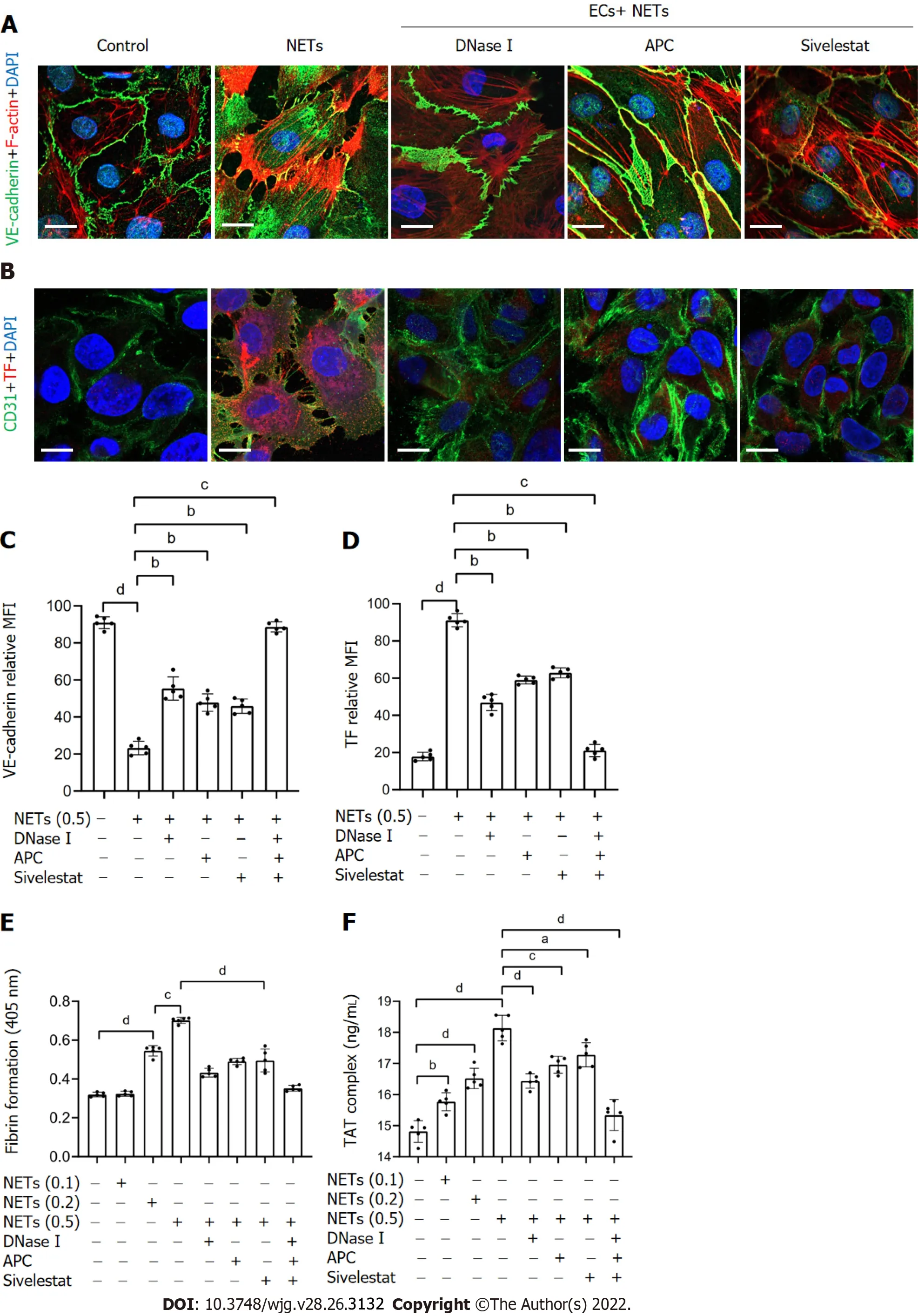
Figure 6 NETs drive hypercoagulation of ECs. A: ECs were cocultured with NETs (μg DNA/mL) or PBS in the presence of DNase I, APC, or sivelestat alone or together for 4 h and analyzed by confocal microscopy. The intercellular junctions of ECs were stained with VE-cadherin and phalloidin. Magnification 63×; scale bars: 10 μm. Red-phalloidin, Green-VE, and Blue-DAPI; B: EC activation was stained with CD31 and TF. Magnification 63×; scale bars: 10 μm. Red-TF, Green-CD31,and Blue-DAPI; C and D: VE-cadherin expression and TF expression on ECs were detected by confocal microscopy and analyzed with ImageJ software (expression indicated as MFI). MFI was defined as the ratio of total fluorescence intensity to the area; E and F: EC monolayers were stimulated with various concentrations of NETs for 4 h, followed by determination of fibrin formation by turbidity measurement at 405 nm, and the TAT complex level was detected by ELISA. All values are the mean ± SD. aP < 0.05; bP < 0.01; cP < 0.001; dP < 0.0001. NETs: Neutrophil extracellular traps; ECs: Endothelial cells; DNase I: Deoxyribonuclease I; APC: Activated protein C; TF: Tissue factor; TAT: Thrombin-antithrombin; MFI: Mean fluorescence intensity.
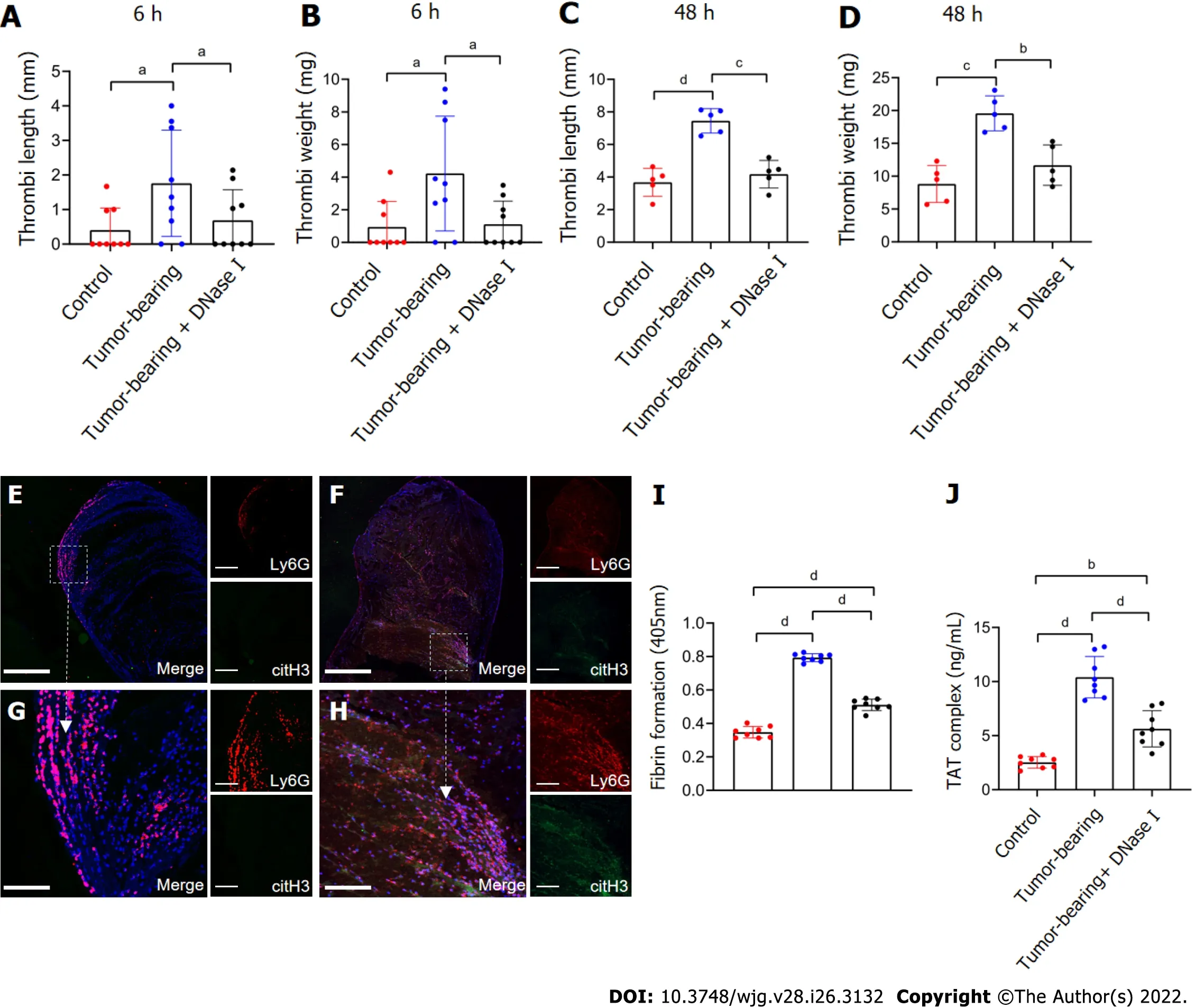
Figure 7 Tumor-bearing mice show a greater ability to form thrombi by inferior vena cava flow restriction. A and B: The values for weight and length of thrombi present in control, tumor-bearing mice, DNase I infused tumor-bearing mice at 6 h after surgery. Each group, n = 9; C and D: Values for weight and length of thrombi present in mice at 48 h after surgery. Each group, n = 5; E and F: Confocal imaging of thrombi derived from control mice and tumor-bearing mice with Ly6G and citH3 staining. Magnification 10×; scale bars: 200 μm. Red-Ly6G, Green-citH3, and Blue-DAPI; G and H: Magnified (40×) part of thrombi derived from control mice and tumor-bearing mice. Scale bars: 50 μm. Red-Ly6G, Green-citH3, and Blue-DAPI; I and J: Fibrin formation levels in the plasma of control, tumorbearing mice, or DNase-I-infused tumor-bearing mice were detected by turbidity measurement at 405 nm, and TAT complex levels were detected by ELISA. Each group, n = 8. All values are the mean ± SD. aP < 0.05; bP < 0.01; cP < 0.001; dP < 0.0001. DNase I: Deoxyribonuclease I; TAT: Thrombin-antithrombin.
In this study, we found that the levels of plasma NET markers and citH3-positive neutrophils were significantly higher in GC patients than in HDs. Expression of NETs decreased significantly after resection of GC tissues. Neutrophil infiltration and NET formation were upregulated in tumor tissues,relative to adjacent paratumor tissues of the same GC patient. The levels of serum D-dimer were positively correlated with tumor TNM stage, consistent with previous findings[48]. These findings suggest that the expression of NETs promotes GC development and thrombosis in the same group of patients.
It has been reported that hepatocellular cancer and hypoxic CM stimulate the production of NETs by neutrophils[44]. However, the relationship between GC cells and neutrophils is poorly understood. Our experiments demonstrated that both metastatic and nonmetastatic GC cancer cells directly stimulated production of NETs from neutrophils, contrary to GES-1 cells. CM of GC cells, but not that of GES-1,also induced the formation of neutrophil-related NETs. This suggests that NET formation is also mediated by factors secreted by GC cells. Inflammatory cytokines, such as interleukin (IL)-1β, IL-6, IL-8,and tumor necrosis factor-, as well as damage-associated molecular patterns, all overexpressed in tumor microenvironments, stimulate neutrophils to release NETs[49,50]. Although IL-8 is the cytokine with the highest overexpression in GC patients, whether it is the main mediator of NET formation and the subsequent underlying mechanism in GC remain to be validated.
A recent study showed that NETs promote thrombosis by activating and promoting the adhesion of platelets in the venous walls of pancreatic cancer patients[22]. Activated platelets express PS on their surface membranes[51]. Additionally, P-selectin expression on the surface membrane of platelets is also associated with thrombosis[52]. In this study, we found that compared to HDs, PS and P-selectin expression on platelets was significantly higher in GC patients, particularly those with stage III/IV GC.NET treatment upregulated PS and P-selectin expression on platelets. Additionally, NETs stimulated the adhesion of normal platelets on glass slides. However, even though DNase I treatment modulated this phenomenon, some platelets still adhered to the glass slides, suggesting that other secretory factors participated in the adhesion property.
Previous studies have shown that histones in NETs promote thrombosis in colorectal cancer patients[53]. NE is another most abundant protein that binds NETs. Although this potent protein stimulates tumor progression bothin vitroandin vivo, the mechanisms underlying NE-mediated cancer-associated thrombosis remain to be clarified. A recent study on DVT using mouse models showed that NE deficiency or NE inhibition alone does not completely inhibit DVT[54]. In this study, we found that hypercoagulation of platelets was not completely mediated by NET DNA, but also by other secretory proteins in the NETs, such as histones and NE. Consequently, DNase I treatment of NETs had no complete effect on hypercoagulation of platelets. However, a combination of DNase I, APC and sivelestat treatment almost completely inhibited hypercoagulation of platelets. Although sivelestat did not show a strong antihypercoagulation effect similar to DNase I, it nonetheless modulated the activation and adhesion of platelets. This demonstrated that histones and NE also participate in the activation and adhesion of platelets.
DVT can be triggered by injury to vascular ECs. Previous studies have shown that under certain malignancies, NETs can induce dysfunction and apoptosis of ECs[35]. In patients with chronic pancreatic disease and pancreatic cancer, NETs exert their cytotoxicity against ECsviaintercellular cell adhesion molecule-1 and vascular cell adhesion molecule-1 expression[26]. Recent studies have shown that treatment of ECs with NETs derived from patients with colorectal cancer promotes and enhances the production of fibrin and the corresponding coagulation[53]. In this study, we found that NET treatment inhibited the secretion of intercellular junctions in ECs and promoted hypercoagulation of platelets by upregulating TF expression. Moreover, NET treatment upregulated the expression of PS on ECs. ECs activated by NETs significantly increased the level of TAT complexes and fibrin generation in the plasma of HDs. Given that a combination of DNase I, APC and sivelestat treatment completely inhibited hypercoagulation, the process was regulated through numerous mechanisms. These findings strongly suggest that NETs contribute to GC-associated thrombosis.
In the late stage of murine mammary tumor models, thrombi were found in lung vessels, and NETs accumulated, indicating that cancer-induced NETs contribute to the cancer-associated thrombosis[19].Here, we demonstrated that GC-bearing mice have a greater ability to form thrombi than control mice have and that NETs were abundantly present in the thrombi of tumor-bearing mouse IVC stenosis models. Most of this response can be blocked by DNase I treatment, which was similar to previous studies. In DVT models, flow restriction of the IVC may result in a hypoxic microenvironment to recruit neutrophils and stimulate NET release. In addition, cancer cells often secrete more inflammatory factors,which aggravate the recruitment of neutrophils to form NETs under a hypoxic conditions[50].Therefore, neutrophils are exposed to two major triggers of NET release: A tumor hypoxic environment and IVC flow restriction, which then participate in the development of thrombi in GC.
CONCLUSlON
Our findings demonstrate that GC cells can directly induce NET formation, which in turn strongly increases the risks of VTE development bothin vitroandin vivo. In addition, we found that not only NET DNA but also histones and NE participate in the development of cancer-associated thrombosis.Accordingly, NETs are potential therapeutic targets against VTE in GC patients.
ARTlCLE HlGHLlGHTS

Research objectives
The present study aimed to investigate the effect of NETs on the development of cancer-associated thrombosis in GC patients.
Research methods
The levels of NETs in blood and tissue samples of patients were analyzed by ELISA, flow cytometry,and immunofluorescence staining. NET generation and hypercoagulation of platelets and endothelial cells (ECs)in vitrowere observed by immunofluorescence staining. NET PCA was determined by fibrin formation and thrombin-antithrombin complex assays. Thrombosisin vivowas measured in a murine model induced by flow stenosis in the inferior vena cava (IVC).
Research results
NETs were likely to form in blood and tissue samples of GC patients compared with healthy individuals.In vitrostudies showed that GC cells and their conditioned medium, but not gastric mucosal epithelial cells, can stimulate NET release from neutrophils. Furthermore, NETs induced hypercoagulable state of platelets and ECs. In a model of IVC stenosis, tumor-bearing mice showed a stronger ability to form thrombi, and NETs abundantly accumulated in the thrombi of tumor-bearing mice compared with control mice. Notably, the combination of deoxyribonuclease, activated protein C,and sivelestat markedly abolished the PCA of NETs.
Research conclusions
Our findings demonstrate that GC-induced NETs strongly increase the risk of VTE development bothin vitroandin vivo. NETs are potential therapeutic targets in the prevention and treatment of VTE in GC patients.
Research perspectives
The treatment strategies can consider the combination of traditional anticoagulant drugs and NETs inhibiting drugs, so as to reduce the risk of cancer associated thrombosis in patients with GC and improve the clinical treatment effect.
FOOTNOTES
Author contributions:Zou XM and Li JC designed the study, completed the experiments, and drafted the manuscript;Yang SF, Zhao TQ, and Jin JQ collected the patient clinical data and performed part of the experiments; Zhu L, Li CJ,and Chen CY participated in the animal experiments; Yang H and Zhang AG performed the statistical analysis; all
authors have read and approved the final manuscript.
Supported byNational Natural Science Foundation of China, No. 81672355.
lnstitutional review board statement:The study was reviewed and approved by the ethics committee of the Second Affiliated Hospital of Harbin Medical University (No. KY2016-032).
lnstitutional animal care and use committee statement:All animal experiments in this study were approved by the ethics committee of the Second Affiliated Hospital of Harbin Medical University (No. KY2016-032).
Conflict-of-interest statement:The authors declare no conflict of interest for this manuscript.
Data sharing statement:No additional data are available.
ARRlVE guidelines statement:The authors have read the ARRIVE guidelines, and the manuscript was prepared and revised according to the ARRIVE guidelines.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:China
ORClD number:Jia-Cheng Li 0000-0002-0467-7225; Xiao-Ming Zou 0000-0002-9689-5026; Shi-Feng Yang 0000-0002-4395-6753; Jia-Qi Jin 0000-0002-7635-5452; Lei Zhu 0000-0002-7495-606X; Chang-Jian Li 0000-0003-0441-0368; Hao Yang 0000-0003-0952-3345; An-Ge Zhang 0000-0003-4772-1254; Tian-Qi Zhao 0000-0002-1938-1419; Chong-Yan Chen 0000-0003-2749-4891.
S-Editor:Fan JR
L-Editor:Kerr C Wang TQ
P-Editor:Fan JR
猜你喜欢
建材发展导向(2021年10期)2021-07-16 07:13:40
高技术通讯(2021年3期)2021-06-09 06:57:24
电站辅机(2017年3期)2018-01-31 01:46:44
电测与仪表(2017年24期)2017-12-19 05:15:16
北京航空航天大学学报(2017年12期)2017-04-23 08:31:39
时代农机(2016年6期)2016-12-01 04:07:27
现代工业经济和信息化(2016年4期)2016-05-17 05:35:43
疯狂英语(双语世界)(2016年3期)2016-02-27 10:11:55
管理现代化(2016年5期)2016-01-23 02:10:11
安徽医药(2014年4期)2014-03-20 13:13:04
 World Journal of Gastroenterology2022年26期
World Journal of Gastroenterology2022年26期
- World Journal of Gastroenterology的其它文章
- Intestinal virome: An important research direction for alcoholic and nonalcoholic liver diseases
- Intestinal inflammation and the microbiota: Beyond diversity
- Epidemiological and clinical aspects of hepatitis B virus infection in Italy over the last 50 years
- Alterations of autophagic and innate immune responses by the Crohn’s disease-associated ATG16L1 mutation
- Advances in the imaging of gastroenteropancreatic neuroendocrine neoplasms
- Divergent trajectories of lean vs obese non-alcoholic steatohepatitis patients from listing to post-transplant:A retrospective cohort study