
聚富马酸酯类交联聚合物的合成及热稳定性能
2022-07-29 02:48:34李文遥张格青王甲威
纺织高校基础科学学报 2022年2期
王 亮,李文遥,张格青,王甲威,董 璐
(西安工程大学 材料工程学院,陕西 西安 710048)
0 引 言
聚富马酸酯是目前富有应用潜力的一种可注射、原位交联及可降解吸收的新型骨科生物医用材料。由于分子中含有富马酸碳碳(C=C)双键结构[1],其不饱和碳碳双键与两端的酯键羰基(C=O)形成共轭结构,使得双键的电子云密度降低,具备更高的反应能力[2],其结构中存在很多活性功能位点,易与多种单体聚合或交联固化,形成具有一定强度的交联网络或固化体。在过去一段时期,人们对聚富马酸丙二醇酯(PPF)及与其他交联剂形成的PPF 基交联共聚物的体内外性能进行了广泛研究,并取得了良好的结果[3-4]。首先,PPF 材料具有良好的生物相容性[5-6]和无毒性[7],且作为一种聚酯材料在体内可以逐渐降解,降解产物为富马酸和丙二醇,可通过正常的新陈代谢排出体外[8]。其次,PPF基交联固化体具有较高的机械强度,在承力骨缺损修复方面展现出良好的应用潜力。目前关于PPF的制备方法有很多,按照合成过程大体可分为一步反应法与两步反应法。一步反应法一般以富马酸或富马酸酯为原料,在一定条件下将其与丙二醇缩合而得到PPF。但一步反应法主要问题在于产物中含有相当比例的副产品,纯度不高[9]。近来,学者们普遍使用两步反应法制备PPF,主要以富马酸二乙酯与丙二醇为初始原料,以富马酸-2-羟基丙酯为中间产物,然后通过酯化聚合反应得到PPF[10]。然而,制备医用级PPF,这些方法仍存在纯度低、分子量范围窄等问题,尤其分子量是影响PPF材料强度、降解速率及黏性等重要性能的关键参数。Kasper 等对两步反应法做了改进,特别对其产物纯化、反应体系水排出方法等提出了详细的实验方案,获得了500~4 000 Da宽分子量范围的高纯PPF,可满足生物医用复合材料要求[1]。聚合物交联固化的方式有很多种,例如热固化、光固化等[11-14]。在2004年5月召开的北美辐射固化国际会议上,光固化技术被归纳为具有高效率、适应范围广、经济节能和环境友好等特点。考虑到聚富马酸丙二醇酯共二丙烯酸酯结构中,除了富马酸单元含有的碳碳双键(C=C)外,两端的2个丙烯酸单元另含有2个双键,可在引发剂作用下进行更复杂多变的交联。本文采用两步法制备聚富马酸丙二醇酯和聚富马酸丙二醇酯共二丙烯酸酯,并利用红外光谱对其结构进行表征。同时考察了聚合物网络形成机理及紫外光辐照时间、辐照强度、引发剂用量等参数对固化行为的影响规律,依据红外光谱监测聚合物网络中相关官能团的吸收峰变化,从而评估交联反应发生的情况。此外,通过TG-DSC曲线分析交联固化体形成过程中光固化反应发生的程度、玻璃化转变温度、热焓变及热稳定性能等。
1 实 验
1.1 试剂与仪器
1.1.1 试剂
富马酸二乙酯(阿拉丁试剂上海有限公司);丙二醇(阿拉丁试剂上海有限公司);氯化锌(国药集团化学试剂有限公司);对苯二酚(天津市科密欧化学试剂有限公司);富马酰氯(北京百灵威科技有限公司);丙烯酰氯(萨恩化学技术上海有限公司);无水碳酸钾(国药集团化学试剂有限公司);三乙胺(阿拉丁试剂上海有限公司);引发剂(西格玛奥德里奇化学试剂);二氯甲烷(天津市富宇精细化工有限公司)。以上均为分析纯。
1.1.2 仪器
Nicolet is50 型傅里叶变换红外光谱仪(美国赛默飞世尔尼高力公司),BUV-6B 型紫外光固化设备(广州市邦沃电子科技有限公司),TGA209C 型热重分析仪(德国耐驰仪器制造有限公司),DSC200PC型差示扫描量热仪(德国耐驰仪器制造有限公司)。
1.2 制备与测试
1.2.1 聚富马酸丙二醇酯的合成
将250 mmol 富马酸二乙酯、750 mmol 丙二醇、2.5 mmol 氯化锌、0.25 mmol 对苯二酚置入反应釜中,通入高纯氮气和回流冷凝水,机械搅拌。体系程序升温如下:待反应体系溶液的温度达到110 ℃,保持0.5 h 不变;升高体系温度至120 ℃,保持0.5 h 不变;最后将反应体系温度提高至140 ℃,反应18 h。
待体系温度降至室温后,再次向反应釜中加入2.5 mmol氯化锌和0.25 mmol对苯二酚,通入高纯氮气,机械搅拌,待体系溶液温度达到100 ℃,保持恒温0.5 h。之后停止通入氮气,开启抽真空系统,使体系的压强降至5 mmHg。继续加热升温,待反应体系温度升高至110 ℃,保持恒温0.5 h;待温度升至120 ℃,保持恒温0.5 h;最后将反应温度提高至130 ℃,反应8 h。待反应结束后得到亮黄色的目标产物即聚富马酸丙二醇酯。
1.2.2 聚富马酸丙二醇酯共二丙烯酸酯的合成
将225 mmol 碳酸钾和450 mmol 丙二醇溶于150 mL二氯甲烷,后置入反应釜中,通入高纯氮气和回流冷凝水,机械搅拌。待体系温度降至0 ℃时,将150 mmol富马酰氯和50 mL二氯甲烷的混合液缓慢滴加到反应体系中,继续反应12 h。待反应结束后得到淡黄色的中间产物即富马酸-2-羟基丙酯。将其溶于适量体积的二氯甲烷中,室温下保存,待用。
向混合溶液中逐滴加入丙烯酰氯,机械搅拌,控制反应体系温度为0 ℃。其中,中间产物、三乙胺、丙烯酰氯三者的摩尔比为1∶3∶2。待丙烯酰氯滴加完毕,在室温条件下反应12 h,再经过水洗涤、盐水洗涤、碱液洗涤、相分离等提纯处理,得到黄色黏稠状目标产物即聚富马酸丙二醇酯共二丙烯酸酯。
1.2.3 交联固化体的制备
依据聚富马酸丙二醇酯结构中富马酸单元的双键数与聚富马酸丙二醇酯共二丙烯酸酯结构中丙烯酸酯单元双键数的摩尔比,将自制的两类聚合物按不同的双键比率(0.5,1,2)混合后作为基本原料,加入适量光引发剂,在紫外光辐照下,制备得到交联固化体。
1.3 结构表征与性能测试
利用红外光谱监测聚合物交联固化体中相关官能团的吸收峰变化,从而判定交联反应发生的情况。取适量交联固化体和溴化钾粉末于玛瑙研钵中充分研磨,混合均匀后压制成薄片。将薄片装在样品架上并放入样品池内,在4 000~400 cm-1的范围内扫描32 次,得到对应的红外光谱。采用耐驰同步热分析仪,测定交联固化体的热失重曲线,分析样品在加热过程中的热稳定性能,测量气流为N2,升温速度为10 ℃·min-1,升温范围为25~600 ℃。在N2氛围下探究聚合物交联固化体的DSC 曲线,依据固化反应过程中的放热量来监测固化反应程度,升温速度为10 ℃·min-1,升温范围为25~300 ℃。
2 结果与讨论
2.1 红外光谱分析
图1 为聚合物的红外光谱。从聚富马酸丙二醇酯的红外光谱可看出,3 450 cm-1附近归属于羟基(—OH)伸缩振动吸收峰;3 000 cm-1处为碳氢(—CH,—CH2,—CH3)伸缩振动吸收峰;1 760~1 690 cm-1附近处吸收峰归属于羰基(C=O)吸收,其一般为最强峰或次强峰,受与羰基相连的基团效应影响,会向高波数或低波数处移动;1 645 cm-1附近处吸收峰归属于碳碳双键(C=C)伸缩振动吸收峰;在1 456 cm-1处为甲基(—CH3)的伸缩振动吸收峰;1 300~1 050 cm-1处吸收峰归属于碳氧(C—O)吸收。以上特征峰的吸收与预期官能团基本一致,可初步认为成功合成聚富马酸丙二醇酯[15-16]。

图1 聚合物的红外光谱图Fig.1 The infrared spectra of polymers
从聚富马酸丙二醇酯共二丙烯酸酯的红外光谱可看出,羟基(—OH)伸缩振动吸收峰位于3 445~3 441 cm-1;碳碳双键(C=C)伸缩振动吸收峰分别位于3 000 cm-1和1 640 cm-1附近;碳氢(—CH、—CH2、—CH3)伸缩振动吸收峰位于2 986~2 961 cm-1;2 359 cm-1附近的峰为二氧化碳吸收;1 728 cm-1和1 711 cm-1附近处吸收峰归属于羰基(C=O),其一般为最强峰或次强峰,受与羰基相连的基团影响,会向高波数或低波数处移动;甲基(—CH3)的伸缩振动吸收峰归属于1 400 cm-1和1 455 cm-1附近处;1 071 cm-1处吸收峰归属于碳氧(C—O)吸收。理论上聚富马酸丙二醇酯共二丙烯酸酯结构中不含有羟基,而实际图谱中出现比较微弱的吸收峰,这可能是由于目标产物中引入了杂质或测试过程中出现误差等。综上分析可知,聚合物的特征官能团吸收峰同预期一致,可以认为成功合成聚富马酸丙二醇酯共二丙烯酸酯[17-18]。
2.2 交联固化机理
图2 为聚富马酸丙二醇酯与聚富马酸丙二醇酯共二丙烯酸酯发生交联固化的反应机理。

图2 形成聚合物网络的反应机理Fig.2 Reaction mechanism of the formation of polymer networks
聚富马酸丙二醇酯主链重复结构单元中的双键与聚富马酸丙二醇酯共二丙烯酸酯结构中两端的双键发生自由基聚合反应,形成网络结构。当紫外光辐照时,光子与分子发生作用,分子吸收能量,由基态转变为激发态。引发剂分子在直接或间接吸收光能后,从基态跃迁到激发单线态,经系间窜跃至激发三线态;在激发单线态或三线态经历单分子或双分子化学作用后,产生能够引发单体聚合的活性碎片。交联固化反应过程中,一部分处在激发单线态的分子,通过释放荧光,返回到基态。在基态中,电子配对且自旋相反。还有一部分转化为过渡态,此过程无光的吸收和发射,只是一些过高的振动能以热的形式放出,即聚富马酸丙二醇酯与聚富马酸丙二醇酯共二丙烯酸酯在光固化过程中有热量放出。
2.3 交联固化体的制备工艺
2.3.1 辐照时间对固化行为的影响
按照设定的双键比率准确称取聚合物,溶于二氯甲烷中混匀,放置12 h 后减压蒸馏除去溶剂。向混匀物中加入适量的引发剂,超声震荡0.5 h 使体系分散均匀,取出后避光放置待用。将混合物均匀地涂布在溴化钾压片上,在4 000~400 cm-1范围内测定其固化前的红外光谱。保持紫外光的辐照强度(310 mW·cm-2)、光源与试样的距离(10 cm)及引发剂用量(0.3 %)一致,探究不同辐照时间(0、10、30、60、180 s)对交联固化体固化行为的影响。不同辐照时间下交联固化涂层在1 635 cm-1和810 cm-1处的红外光谱如图3所示。
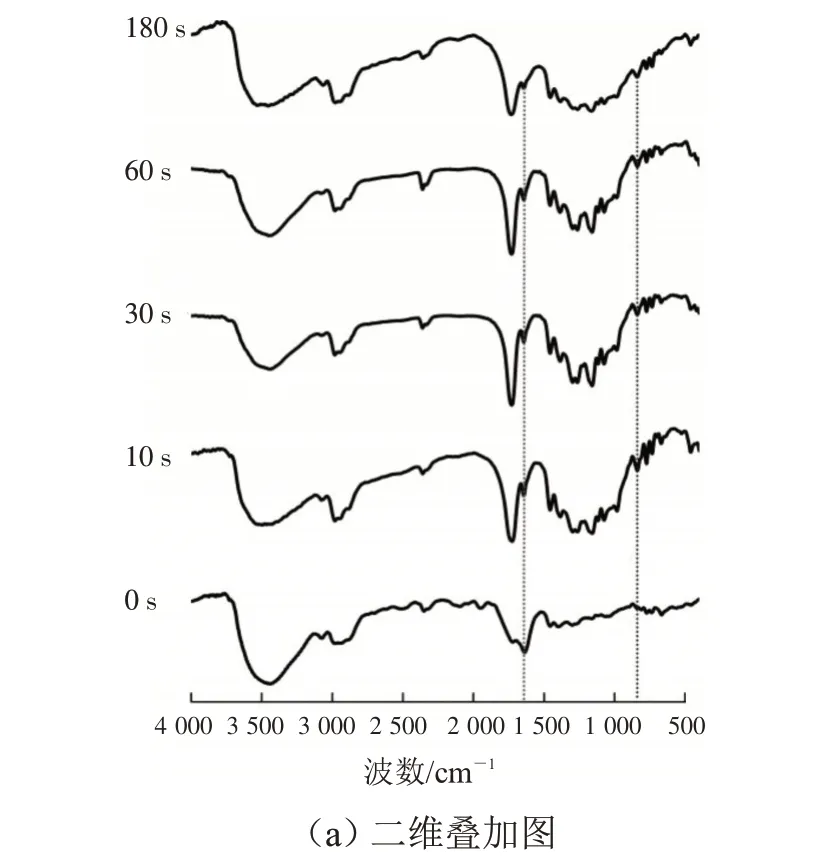

图3 不同辐照时间下固化涂层在1 635 cm-1和810 cm-1处的红外光谱Fig.3 FTIR spectra of UV-curable coating with different irradiation time at 1 635 cm-1 and 810 cm-1
从图3 可看出,随着紫外光辐照时间的增加,红外光谱中1 635 cm-1和810 cm-1处的吸收峰逐渐减弱。当紫外光辐照时间为0 s 时,体系是2 种聚合物的共混物,几乎没有交联反应发生,所以碳碳双键(C=C)吸收峰最强(1 635 cm-1处较为明显)。当紫外光辐照时间为180 s 时,位于1 635 cm-1处红外吸收峰最弱。这是由于紫外光辐照时间增大,聚富马酸丙二醇酯结构中富马酸酯单元和聚富马酸丙二醇酯共二丙烯酸酯结构中丙烯酸酯单元上的碳碳双键(C=C)更多参与交联反应中[19]。
2.3.2 辐照强度对固化行为的影响
保持紫外光辐照时间(180 s)、光源与试样的距离(10 cm)及引发剂用量(0.3%)一致,探究不同紫外光辐照强度(0%、30%、50%、80%、100%)对材料固化行为的影响。依据光固化设备输出功率的换算,其对应的紫外光的辐照强度大小依次为0、93、155、248、310 mW·cm-2。不同辐照强度下交联固化涂层在1 635 cm-1和810 cm-1处红外光谱如图4所示。
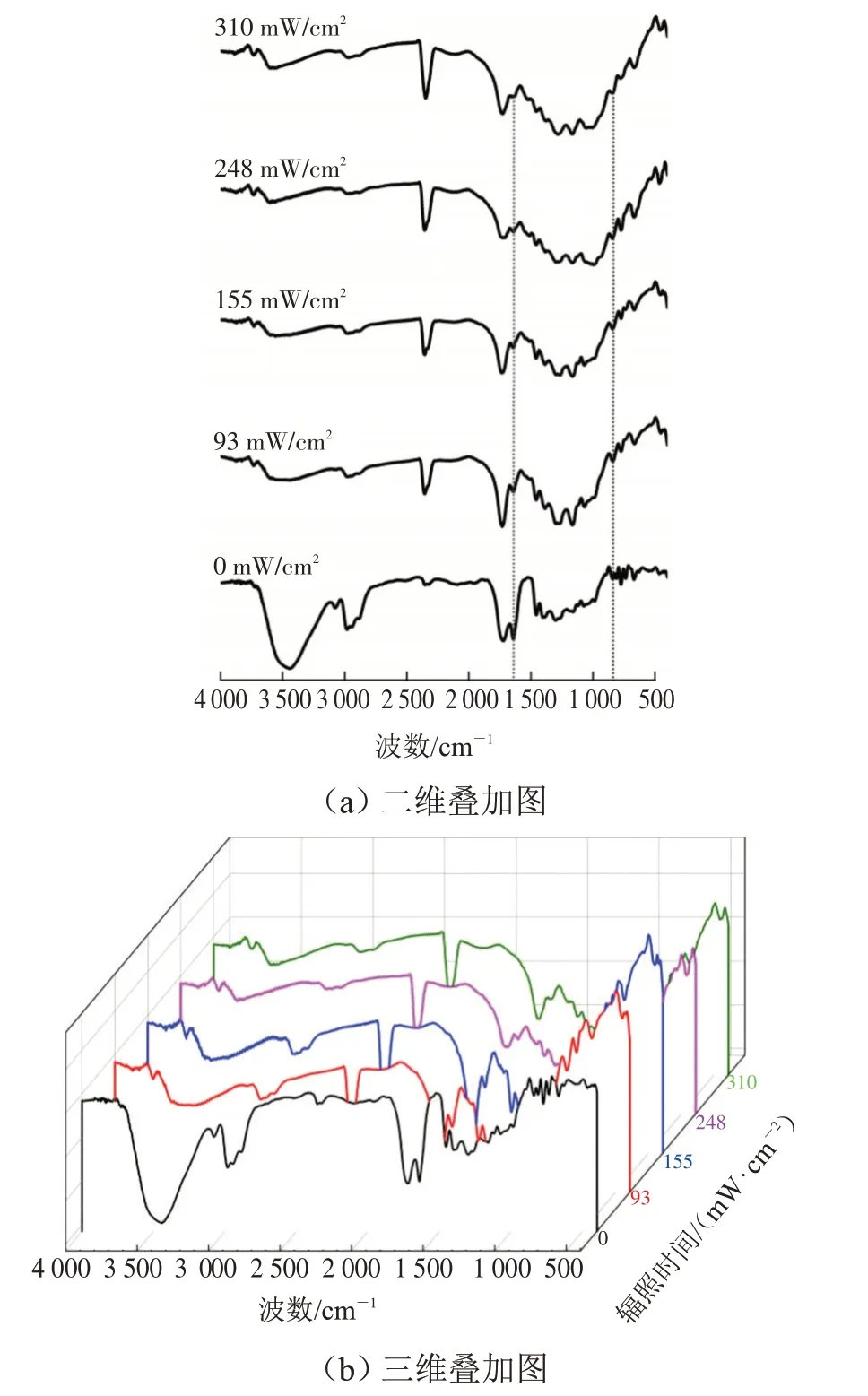
图4 不同辐照强度下固化涂层在1 635 cm-1和810 cm-1处的红外光谱Fig.4 FTIR spectra of UV-curable coating with different irradiation intensity at 1 635 cm-1 and 810 cm-1
从图4 可看出,随着紫外光辐照强度的增加,1 635 cm-1和810 cm-1处的吸收峰逐渐减弱。当试样没有被紫外光辐照时,体系是聚合物的共混物,几乎未发生交联反应,故碳碳双键(C=C)红外吸收峰最强(1 635 cm-1处较为明显)。当紫外光辐照强度为310 mW·cm-2时,1 635 cm-1和810 cm-1处的吸收峰最弱,这是由于紫外光辐照强度增大,加速了聚合物的交联反应的发生,消耗了更多的碳碳双键[19]。
2.3.3 引发剂用量对固化行为的影响
保持紫外光辐照时间(180 s)、辐照强度(310 mW·cm-2)及光源与试样的距离(10 cm)一致,探究引发剂用量(0.1%、0.2%、0.3%、0.4%)对材料固化行为的影响。不同引发剂用量下交联固化涂层在1 635 cm-1和810 cm-1处红外光谱如图5所示。
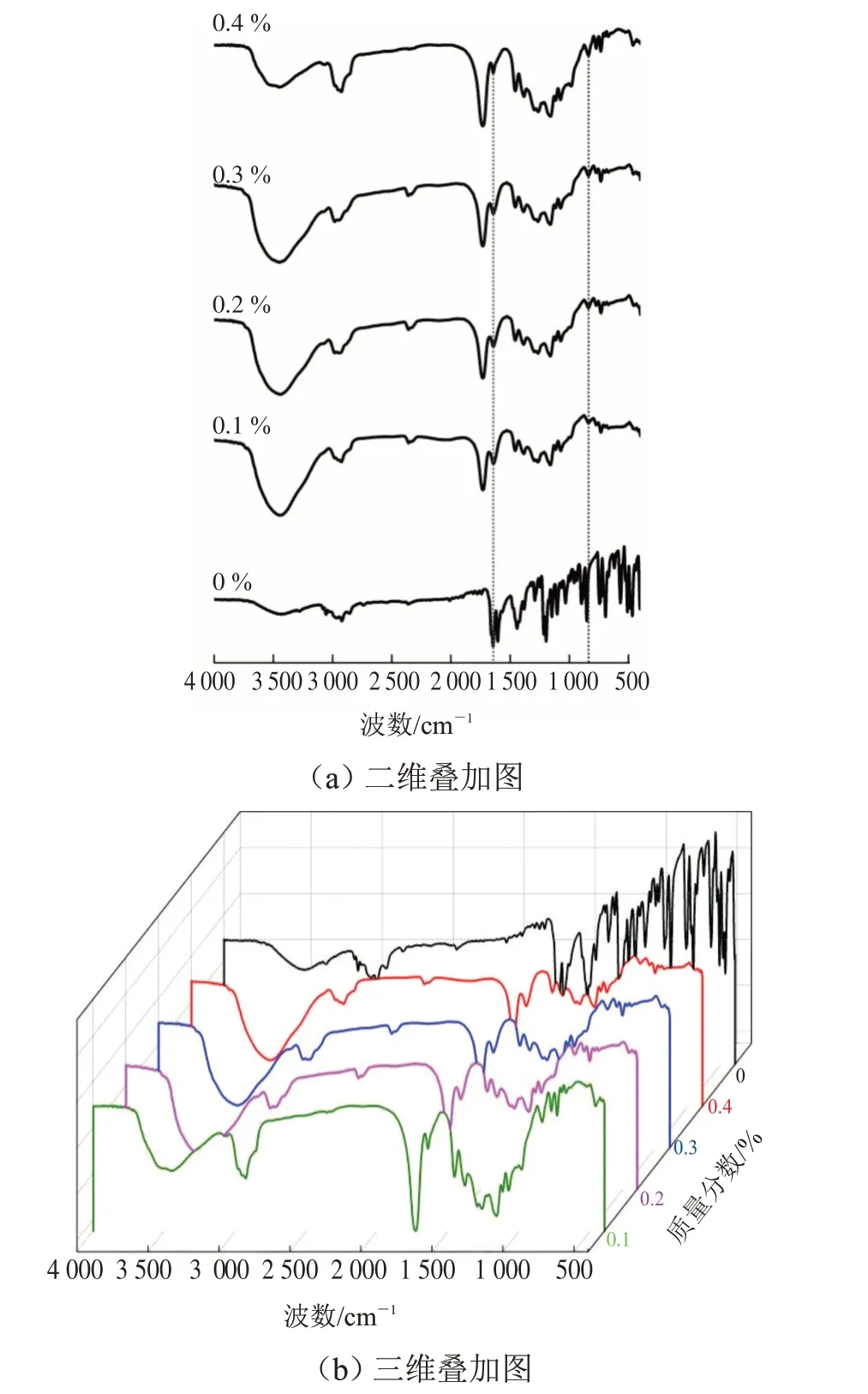
图5 不同引发剂用量下固化涂层在1 635 cm-1和810 cm-1处的红外光谱Fig.5 FTIR spectra of UV-curable coating with different initiator dosage at 1 635 cm-1 and 810 cm-1
从图5 可看出,随着引发剂用量的增加,1 635 cm-1和810 cm-1处的吸收峰逐渐减弱,表明碳碳双键(C=C)更多地参与交联反应。在紫外光固化体系中,引发剂在紫外光辐照下,自由基是均匀形成的。随着引发剂用量的增加,自由基浓度增大,其交联固化速率变快。由于光引发剂对紫外光的吸收,使其穿透能力减弱,致使材料固化不均匀,甚至阻碍了底层固化行为。当引发剂的质量分数为0.4%时,聚合物的交联速率较快,固化过程中因局部过热产生气泡,从而影响了试样的质量。因此,引发剂用量为0.3%较为适宜。
2.4 热稳定性分析
交联固化体的热失重曲线和DSC 曲线如图6所示,升温速率10 ℃/min。热失重曲线得到的实验值同理论计算值基本一致。

图6 3种双键比率交联固化体的热失重曲线及DSC曲线Fig.6 Thermogravimetric curves and DSC curves of cross-linked network with three kinds of doublebond ratios
由图6可看出,3种双键比率的交联固化体都经历一次明显的失重过程,开始分解的温度为271.60oC~287.98oC,于365.29oC~368.56oC 范围出现了一个明显吸热峰,其最大质量损失率为79.55%~81.55%。当聚合物的双键比率为0.5时,交联固化体加热至287.98oC 时没有出现明显的质量损失,表明交联固化体在这个温度范围内可以保持结构完整性。交联固化体在287.98oC~483.28oC 时经历一个主要的质量损失阶段,其中在368.56oC 时出现一个明显的吸热峰,总体质量损失率接近81.55%。当聚合物的双键比率为1 时,交联固化体在285.26oC~473.47oC 出现一次明显的失重,总体质量损失率达79.55%,且吸热峰出现在367.36oC处。此外,当聚合物的双键比率为2 时,通过TG 曲线可以观察到一个明显的质量损失,在271.60oC~475.39oC 范围,其中365.29oC 处出现吸热峰,质量损失率为80.09%。综合分析可知,双键比率对交联固化体的热稳定性能有着重要的影响,当双键比率为0.5 时,交联固化体的热稳定性能较好。
由DSC 曲线可知,当双键比率为0.5 时,在166.61oC~194.72oC时发生了明显反应,玻璃化转变温度为177.92oC,释放热量为14.69 J·g-1;双键比率为1 时,在139.28oC~160.44oC 时发生了明显反应,玻璃化转变温度为154.36oC,释放热量为3.10 J·g-1;双键比率为2 时,在温度区间为148.81oC~172.62oC时发生了明显反应,玻璃化转变温度为161.44oC,释放热量为6.26 J·g-1。结果表明,当交联固化体的双键比率为0.5 时,材料的玻璃化转变温度高,固化反应释放的热量大,其固化反应速率较慢。这是因为丙烯酸酯基团可以提高聚合物网络的刚性,从而阻碍了交联反应的发生。换言之,交联固化体中的双键比率会影响其交联密度和玻璃化转变温度,这是因为玻璃化温度与链的柔韧性、交联密度等存在依赖关系[20-21]。
3 结 论
1)成功合成了聚富马酸丙二醇酯和聚富马酸丙二醇酯共二丙烯酸酯,并通过光固化手段获得不同双键比率的聚富马酸丙二醇酯/聚富马酸丙二醇酯共二丙烯酸酯交联固化体。
2)聚富马酸丙二醇酯主链重复结构单元中的双键与聚富马酸丙二醇酯共二丙烯酸酯结构中两端的双键发生自由基聚合反应,形成交联网络结构。交联固化体的较适宜的固化工艺为:紫外光辐照时间120 s,紫外光辐照强度248 mW·cm-2,引发剂的用量0.3%。
3)交联固化体开始分解的温度为271.60oC~287.98oC,于365.29oC~368.56oC 范围出现了一个明显吸热峰,其最大质量损失率为79.55%~81.55%。交联固化体的玻璃化转变温度为154.36oC~177.92oC,与之对应的焓变变化为-3.10 J·g-1~-14.69 J·g-1。
猜你喜欢
石油化工技术与经济(2023年6期)2024-01-31 05:12:04
油气·石油与天然气科学(2021年12期)2021-12-11 01:43:23
皮肤病与性病(2021年3期)2021-07-30 08:08:48
皮肤病与性病(2021年3期)2021-07-30 08:08:02
洛阳理工学院学报(自然科学版)(2020年1期)2020-05-15 09:24:02
分析化学(2017年12期)2017-12-25 12:43:03
国外医药(抗生素分册)(2015年3期)2015-07-12 12:28:27
杭州师范大学学报(自然科学版)(2015年5期)2015-03-20 01:13:42
婚姻与家庭·性情读本(2014年12期)2014-12-17 01:17:16
化工技术与开发(2013年1期)2013-09-27 06:43:28
