
X-linked osteogenesis imperfecta accompanied by patent ductus arteriosus:a case with a novel splice variant in PLS3
2022-07-29 10:17:42ChuangQiuQiWeiLiLuZhangXiaoLiangLiu
World Journal of Pediatrics 2022年7期
Chuang Qiu ·Qi-Wei Li ·Lu Zhang ·Xiao-Liang Liu
Osteogenesis imperfecta (OI),also known as brittle bone disease,is characterized by low bone mass and bone fragility,which varies in severity from asymptomatic to perinatal lethality [1].Most patients with OI experience recurrent bone fractures with or without short stature,facial dysmorphism,and skeletal deformities.The accompanying extraskeletal manifestations include blue sclerae,joint laxity,dentinogenesis imperfecta,and hearing impairment.Approximately 85—90% of patients with OI have mutations in eitherCOL1A1
orCOL1A2
,which elicit defects in type I collagen.Recently,an X-linked subtype of OI that is unrelated to collagen was identified and was attributed to mutations in the plastin 3 gene (PLS3
) [2].PLS3
(OMIM 300131) is a highly conserved gene comprising 16 exons located on chromosome Xq23.PLS3
encodes a 630-amino acid-long protein that participates in the dynamic assembly and disassembly of actin bundles in the cytoskeleton [3].PLS3 is ubiquitously expressed in solid tissues and is involved in multiple processes,including cell migration,endocytosis,DNA repair,and membrane trafficking [4].Intriguingly,the currently identifiedPLS3
mutations seem to mainly influence the skeleton,which have been associated with X-linked,child-onset,primary osteoporosis and bone fragility.More studies are needed to expand the mutation spectrum as well as to improve our understanding of the phenotypic diversity and therapeutic options forPLS3-
associated OI.Here,we report the clinical characteristics of a boy diagnosed with OI and patent ductus arteriosus (PDA).A novel splice variant ofPLS3
was identified that elicits loss of function of PLS3 via altered splicing and nonsensemediated mRNA decay (NMD).The patient was followed up for 2 years to evaluate the efficiency of bisphosphonate treatment.The proband was from the outpatient clinic of Shengjing Hospital of China Medical University.The serum Ca,P,Mg,and 25-hydroxyvitamin D (25OHD) levels were determined by an automated chemical analyzer (Siemens,Germany).Bone-specific alkaline phosphatase (BAP) and β-cross linked C-telopeptide of type I collagen (β-CTX) were determined by an automated electrochemiluminescence system(Roche Diagnostics,Switzerland).Bone mineral density(BMD) was measured by dual-energy X-ray absorptiometry(DXA,GE Healthcare,Madison,WI,USA).
Written informed consent was obtained before wholeexome sequencing (WES) using biotinylated capture probes(MyGenostics,Baltimore,MD,USA).The mutation was confirmed by Sanger sequencing.In silico prediction was performed with NNSPLICE0.9 (http://www.fruitfly.org/).
Reverse transcription-polymerase chain reaction (RTPCR) was carried out on peripheral blood lymphocytes with primers 5′-TCC GTA ACT GGA TGA ACT CT-3′ and 5′-TGA TGT CGT CAT TGG CTT T′ forPLS3
(NM_001136025.5),and 5′-AAC TGG GAC GAC ATG GAG AAA-3′ and 5′-TAG CAC AGC CTG GAT AGC AAC-3′ for β-actin(NM_001101.5).Western blotting was performed using primary anti-PLS3 antibody (1:1000;Abcam) or anti-actin antibody (1:1000;Abcam,MA,USA).The proband was a 9-year-old Chinese boy who was referred for genetic counseling.He was born with PDA and received surgical repair at 3 years old.Both cognitive and motor development were unremarkable.He had experienced low-energy hairline fractures three times:the first occurred on the left humerus at the age of 13 months,the second was on the right humerus at the age of 2 years,and the third was on the right fifth finger at the age of 8 years.His parents were healthy and non-consanguineous,and no family members had fracture histories.His height and weight were 132.4 cm(Z
-score —0.55) and 31.3 kg (Z
-score+0.19),respectively.He had blue sclerae,joint hyperlaxity,and left fifth finger camptodactyly.No abnormalities were found in gait pattern,muscle strength,dentinogenesis,or hearing.Skeletal radiography (Fig.1 a,c,e,g) demonstrated a thin bone cortex,the“S”form of scoliosis,thoracic kyphosis,lumbar lordosis,slight wedging of several vertebrae,and a bend in the second phalanx of the left fifth finger.No obvious vertebrate compression fractures or long bone deformities were observed.Serum Ca,P,Mg,and 25OHD levels were within normal ranges.The serum levels of the bone turnover markers BAP(147.45 μg/L,reference:< 20.1 μg/L) and β-CTX (1.31 μg/mL,reference:0.26-0.512 μg/mL) were much higher than normal (Fig.1 h,i).The total body BMD (0.674 g/cm 2,Z
-score -3.04) and total body less head BMD (0.574 g/cm 2,Z-score -2.35) were far below the normal levels (Fig.1 j).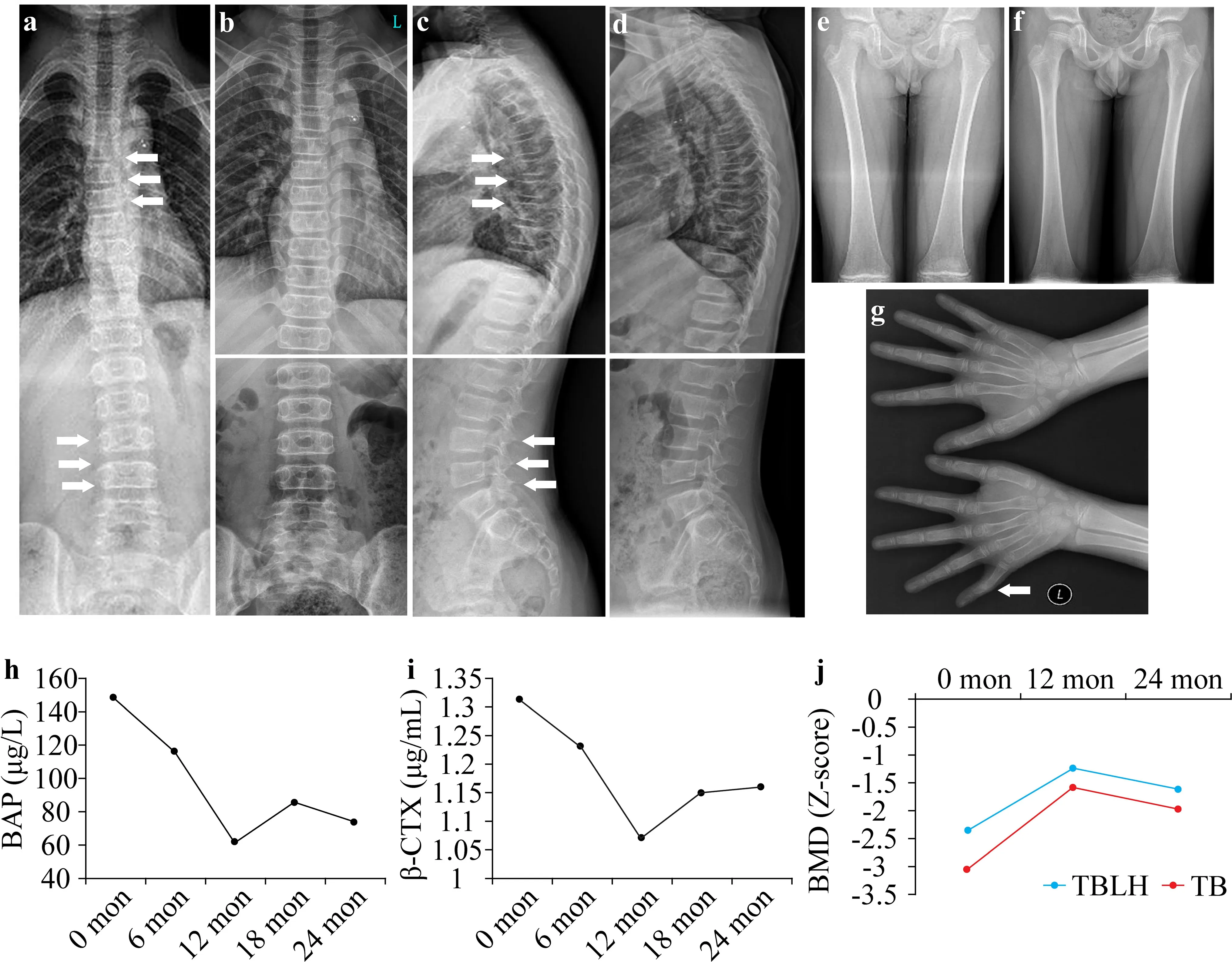
Fig.1 Radiographic and laboratory manifestations of the patient.Radiographs of the spine and extremities before (a,c,e,g) and after(b,d,f) 2 years of bisphosphonate treatment are shown.The“S”form of scoliosis;thoracic kyphosis;lumbar lordosis;slight wedging in T5,T7,T8,and L4;and a bend in the second phalanx of the left fifth finger are indicated by white arrows.The bone turnover markers of serum bone-specific alkaline phosphatase (BAP) (h) and β-cross linked C-telopeptide of type I collagen (β-CTX) (i) recorded during the 2 years of follow-up.The Z-scores for bone mineral density(BMD) were evaluated during the follow-up (j).TB total body,TBLH total body less head
WES for the proband identified a novel hemizygous c.1262+1G > A variant inPLS3
(Fig.2 a).Sanger sequencing confirmed the variant to be de novo,which was not detected in either parent (Fig.2 b).In silico prediction(Fig.2 c) showed that the mutation causes disruption of the original donor splice site in intron 11 and adoption of an alternative donor site at c.1262+192 with a splice score of 0.94.The transcription read-through might encounter a premature termination codon (PTC),eliciting a 422-amino acid truncation or null expression by PTC-induced NMD.RTPCR analysis detected a transcript ofPLS3
in the age-and sex-matched healthy control but not in the patient (Fig.2 d).Western blotting (Fig.2 e) did not show full-length plastin 3(approximately 70 KD) or any truncated forms in the patient.Therefore,we concluded the pathogenic mechanism of the c.1262+1G > A variant to be loss of function via altered splicing and PTC-induced NMD.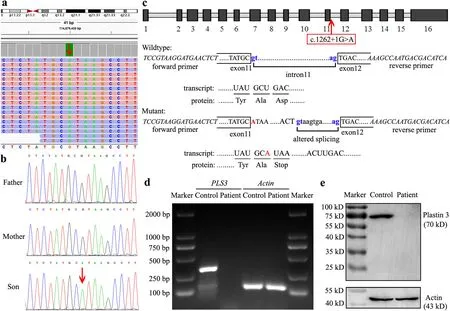
Fig.2 Sequencing and expression analyses of PLS3.A hemizygous c.1262+1G > A variant of PLS3 was found in the patient using whole-exome sequencing (a),and was confirmed to be de novo using Sanger sequencing (b).The variant is indicated by red arrows.Schematic representation and splicing prediction of the wild-type and mutant PLS3 sequences using NNSPLICE0.9 (http://www.fruitfly.org/) (c).The variant in intron 11 is indicated in red and the putative splice sites are indicated in blue letters.The transcription of PLS3 was analyzed using RT-PCR (d),showing an amplified 363-bp transcript encompassing exon 11 to exon 14 in the healthy control but not in the patient.The β-actin transcript was used as the internal control.The protein expression of plastin 3 was detected by western blotting(e),showing expression of neither full-length (approximately 70 KD)nor any truncated forms in the patient.The actin protein (approximately 43 KD) was used as the internal control
The patient was provided with bisphosphonate treatment and was followed up for 2 years.No new fracture occurred,and the scoliosis and kyphosis were significantly corrected (Fig.1 b,d,f).Serum Ca,P,Mg,and 25OHD levels remained within reference ranges continuously.The levels of the bone turnover markers significantly decreased at the first year of follow-up but slightly rebounded the second year(Fig.1 h,i).Moreover,theZ
-scores for BMD improved in the first year but slightly declined during the second year of treatment (Fig.1 j).PLS3
was newly identified in 2013 to be associated with X-linked OI [2].However,the understanding of this rare subtype of OI remains limited.To date,27 pathogenic variants ofPLS3
have been found,including the one reported herein [5,6].The mutation types are variable,including five deletion mutations,five nonsense mutations,four splice mutations,four missense mutations,two insertion mutations,two duplication mutations,as well as five large fragment copy number variations encompassing the complete or partialPLS3
gene.PLS3 protein is structurally constituted by an N-terminal regulatory domain,a flexible linker,and a core consisting of two actin-binding domains [7].Most of the variants have been identified in the actin-binding domains.The present study adds a novel splice variant to the mutation spectrum,resulting in the null expression ofPLS3
in the patient via NMD as the pathogenic mechanism.We have reviewed the clinical features in all reported 46 patients (1 female and 45 males) carryingPLS3
mutations.These patients showed low BMD,low-energy peripheral or vertebrae compression fractures,and common secondary OI features,including blue sclerae,hearing loss,dentinogenesis imperfecta,joint hypermobility,short stature,or facial dysmorphism.There were also some extraskeletal non-OI symptoms in 13 of the male patients [2,6,8— 13] (Table 1).It is noteworthy that PDA was reported in two other patients carrying c.235delT (p.Y79IfsX6) and c.994_995delGA(p.D332X) mutations,respectively [2,8].This case thus represents the third such association ofPLS3
-associated OI with PDA,which suggests a correlation of these two phenotypes.The normal process of ductus arteriosus closure involves regulated cell—matrix interactions between smooth muscle cells,endothelial cells,and the extracellular matrix [14].PLS3 has been shown to play a role in thoracic aortic dissection via endothelial dysfunction [15].Thus,the involvement of PLS3 in ductus arteriosus closure is suspected through inducing vascular cell dysfunction,which deserves further investigation.Table 1 -associated X-linked osteogenesis imperfecta (OI) cases with non-OI symptoms
regulatory domain, actin-binding domains,calponin homology,not available
Treatment againstPLS3
-associated OI with either antiresorptive bisphosphonate [5,9,11,13,16] or osteogenic teriparatide[17,18] has been reported.Overall,bisphosphonate contributes to reshaping compressed vertebral bodies,improvement of vertebral body height,and modification of kyphosis.An obvious increase in BMD and appropriate suppression of bone turnover markers also were observed,especially in the early period of bisphosphonate treatment.Teriparatide treatment resulted in a marginal increase in BMD and a significant increase in the levels of bone turnover markers.However,teriparatide decreased the histomorphometric variables of trabecular osteocyte lacunae size and eroded surface.Our patient undergoing bisphosphonate treatment showed satisfactory correction of scoliosis andkyphosis,a marked increase in BMD,and a decrease in the levels of bone turnover markers in the first year of treatment.Collectively,based on previous findings and the present case,bisphosphonate treatment is recommended forPLS3
-associated OI.It should be noted that long-term antiresorptive therapy may be associated with a risk of atypical femoral fractures,which indeed occurred in an adolescent boy withPLS3
mutation after 7 years of intravenous and 2 years of oral bisphosphonate treatment [19].A hiatus or an alternative teriparatide treatment could be taken into consideration.In conclusion,we have described a rare case of X-linked OI accompanied by PDA.A novel splice variant was identified causing loss of function of PLS3 via NMD.Bisphosphonate was therapeutically beneficial,especially in the first year.These data enrich our understanding about the phenotype diversity,molecular basis,and efficiencies of treatments forPLS3
-associated OI.Acknowledgements
We would like to thank the individuals who participated in this study.Author contributions
CQ:data curation,resources,writing—original draft.QL:investigation,visualization.LZ:methodology,software.XL:conceptualization,funding acquisition,supervision,writing—review and editing.Funding
This study was supported by grants from The National Key Research and Development Program of China (2021YFC1000300,2021YFC1000304) and New Medical Technology and Project of China Medical University (112-3110210717).Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Declarations
Conflict of interest
No financial or non-financial benefits have been received or will be received from any party related directly or indirectly to the subject of this article.Ethics approval
The study was conducted in accordance with the Helsinki Declaration and approved by the Ethics Committee of Shengjing Hospital of China Medical University (2021PS535K).Written informed consent to participate in the study have been obtained from the parent of the patient.Consent for publication
Written consent for publication of the case details together with imaging or videos have been obtained from the parent of the patient. World Journal of Pediatrics2022年7期
World Journal of Pediatrics2022年7期
- World Journal of Pediatrics的其它文章
- Editors
- Information for Readers
- Prevalence of congenital cytomegalovirus infection in preterm,small for gestational age and low birth weight newborns:characteristics and cytokines profile
- Instructions for Authors
- Newborn emergency transport based on the fifth-generation wireless networks and blockchain
- Trends in prevalence and mortality of gastroschisis and omphalocele in the United States from 2010 to 2018