
Natural product-based radiopharmaceuticals:Focus on curcumin and its analogs,flavonoids,and marine peptides
2022-07-22 03:52HendrisWongso
Hendris Wongso
Labeled Compound and Radiometry Division,Center for Applied Nuclear Science and Technology,National Nuclear Energy Agency,Bandung,40132,Indonesia
ABSTRACT
Natural products provide a bountiful supply of pharmacologically relevant precursors for the development of various drug-related molecules,including radiopharmaceuticals.However,current knowledge regarding the importance of natural products in developing new radiopharmaceuticals remains limited.To date,several radionuclides,including gallium-68,technetium-99m,fluorine-18,iodine-131,and iodine-125,have been extensively studied for the synthesis of diagnostic and therapeutic radiopharmaceuticals.The availability of various radiolabeling methods allows the incorporation of these radionuclides into bioactive molecules in a practical and efficient manner.Of the radiolabeling methods,direct radioiodination,radiometal complexation,and halogenation are generally suitable for natural products owing to their simplicity and robustness.This review highlights the pharmacological benefits of curcumin and its analogs,flavonoids,and marine peptides in treating human pathologies and provides a perspective on the potential use of these bioactive compounds as molecular templates for the design and development of new radiopharmaceuticals.Additionally,this review provides insights into the current strategies for labeling natural products with various radionuclides using either direct or indirect methods.
Keywords:
Natural products
Radiopharmaceuticals
Radiolabeling
Curcumin
Flavonoids
Marine peptides
1.Introduction
Radiopharmaceuticals play a substantial role in modern medicine mainly for the diagnosis and treatment of various diseases.To date,the field of nuclear medicine is continuously evolving,as exemplified by the growing number of radiopharmaceutical products covering a wide range of active compounds,including organic molecules,nanoparticles,peptides,proteins,and monoclonal antibodies[1-3].The Society of Nuclear Medicine reported that approximately 20 million nuclear medicine procedures are performed in the United States each year to detect diseases and deliver targeted therapies[4].
Despite the considerable success in developing radiopharmaceuticals for human use,there is still room for improvement.For example,several radiopharmaceuticals have been designed and developed to understand brain pathologies in the neuroscience field.Although the candidates have shown promising results in both in vitro and animal studies,they are often unsuccessful in clinical trials[5].In recent decades,some radiopharmaceuticals have been studied as positron emission tomography(PET)agents(e.g.,[11C]PBB3,[18F]AV1451,[18F]MK-6240,[18F]RO69558948,[18F]PI-2620,and[18F]JNJ311)in neurodegenerative tauopathies to assist with disease staging and to identify the underlying causes of brain diseases.Unfortunately,most of them exhibit stability issues,off-target binding,and lack of specificity and selectivity[6].Moreover,treatment using radiopharmaceuticals can be an effective approach in oncology when other standard therapeutic strategies have failed.Nevertheless,radiotherapy has not become a component of cancer treatment armamentarium in the same way as other approaches.Notably,some radiopharmaceuticals target a pathway that is not involved in promoting the cancer phenotype,resulting in clinical trial failure[7].Hence,the development of radiopharmaceuticals remains an interesting challenge.
It is noteworthy that molecules of synthetic origin essentially dominate the progress in radiopharmaceutical development.In contrast,the development of radiopharmaceuticals from natural products remains largely unexplored.Therefore,further exploration of natural product-based radiopharmaceuticals is required to address some of the limitations associated with synthetic-based radiopharmaceuticals.Although the utilization of natural products as a template for the development of new radiopharmaceuticals is limited,several radiopharmaceuticals derived from natural bioactive molecules have been synthesized and developed and have shown promising applications in the field of nuclear medicine,encouraging drug developers to further explore the potential and feasibility of bioactive constituents from the natural environment.
Although it appears promising,several existing challenges critically hinder or prevent the effective exploration of natural product-based radiopharmaceuticals.For instance,radiopharmaceutical candidates(bioactive precursors)must have a suitable site for labeling with radionuclides[8];however,many natural molecules possess structural complexity that requires molecular modifications to allow the attachment of the radionuclide.In general,the modifications may include the conversion of a specific group into a functional moiety that enables conjugation with a radionuclide,protection and deprotection of nucleophilic group(s)to avoid unwanted side reaction(s),and the addition of a chelator or linker to prevent steric impact on binding to the receptor.Additionally,ensuring access and adequate quantities of bioactive constituents from nature using either isolation methods or synthetic strategies is one of the greatest challenges.The complexities of natural product chemistry are often associated with slowness in isolation and difficulty in structure elucidation.Furthermore,obtaining intellectual property rights of newly designed or several novel drugs derived from natural products can be problematic[9,10].
It is evident that the development of natural product-based radiopharmaceuticals is synthetically challenging.However,opportunities now exist to take full advantage of the diversity of natural products.These challenges maybe mitigated by discovering alternative procedures for well-conserved radiolabeling strategies.Thus,an increase in natural product-based radiopharmaceutical research is expected in the near future.This review highlights the opportunities brought on by natural products to develop new radiopharmaceuticals,the challenges that we still face,and the innovative strategies that can be utilized in the radiosynthesis of natural product-based radiopharmaceuticals.
2.Biological activities of natural products
In the last three billion years,nature has played a major role in the production of bioactive compounds with massive structural and functional diversities that far exceed the current synthetic chemistry capability.As time passes,natural products have been found to be highly beneficial for a variety of purposes for humans.Natural products can interact with multiple molecular targets and,thus,are privileged scaffolds for drug development[9,11,12].This has resulted in an extensive series of compound classes that display diverse biological activities,such as antimicrobial,antidiabetic,anticancer,antioxidant,and anti-inflammatory activities[10].Owing to the vast number of bioactive natural products available,it is difficult to cover all features of all compounds in this review.Therefore,here,only three groups of compounds are discussed:curcumin and its analogs,flavonoids,and marine peptides.These compounds were selected as they show promise as template molecules for the design and development of novel radiopharmaceuticals capable of detecting and/or treating human diseases.More importantly,these molecules possess functional group diversity that allows structural modifications,resulting in suitable precursors for radiopharmaceutical preparations.In particular,the hydroxyl group attached to the aryl moiety of the curcumin structure facilitates direct radioiodination or attachment of a carbon linker.Similarly,the phenol constituent on the structure of flavonoids and amine,amide,carbonyl,and carboxylic acid groups on marine peptides might be useful for the direct or indirect attachment of radionuclides.
2.1.Curcumin and its analogs
Over the past few decades,turmeric(Curcuma longa)has been used as a medicinal herb to treat many health problems,such as cancer,infection,inflammation,neurodegenerative diseases,malaria,various skin diseases,immune diseases,and diabetes[13,14].Turmeric contains the following three essential analogs:curcumin,demethoxycurcumin,and bisdemethoxycurcumin,collectively referred to as curcuminoids[15].Of these analogs,curcumin comprises 0.3%-5.4% of raw turmeric and is the most studied compound,especially for its anticancer properties[16].Curcumin is a symmetrical molecule with two similar-looking aromatic rings that consist of four chemical entities:aryl moieties,which are connected by a carbon linker in the presence of a diketo functional group;two double bonds;and a functional methylene group[14](Fig.1).
Recently,the antibacterial activity of curcumin against Bacillus subtilis(B.subtilis)and Escherichia coli(E.coli)was examined.Silver and gold curcumin nanoparticles showed a maximum zone of inhibition at concentrations of 300μg/mL against B.subtilis and E.coli,respectively[17].In another study,Mody et al.[16]showed that curcuminoids inhibited the growth of Clostridium difficile at concentrations of 4-32 mg/mL without disturbing the major population species of the human gut.Subsequently,the antifungal properties of curcumin were investigated by Martins et al.[18]against 23 fungal strains.In this study,Paracoccidioides brasiliensis isolates were found to be the most vulnerable to curcumin;however,curcumin was the least effective against Aspergillus sp.isolates.
In the past few years,curcumin has been found to exhibit different anticancer activities in multiple in vitro and in vivo studies.Woo et al.[19]demonstrated that curcumin could induce the expression of 15-hydroxyprostaglandin dehydrogenase at both transcriptional and translational levels in normal rat gastric mucosal cells and could,therefore,potentially be used as a chemopreventive agent against inflammation-associated gastric carcinogenesis.San et al.[20]discovered that curcumin could enhance the efficacy of the chemotherapeutic agent suberoylanilide hydroxamic acid in anoikis-resistant cholangiocarcinoma cells.Similarly,Liu et al.[21]demonstrated the role of curcumin in improving cancer drug efficacy.The authors revealed that curcumin might promote the apoptosis of pancreatic cancer cells through the PARP/caspase-3 signaling pathway and increase the pro-apoptotic ability of either gemcitabine or docetaxel.
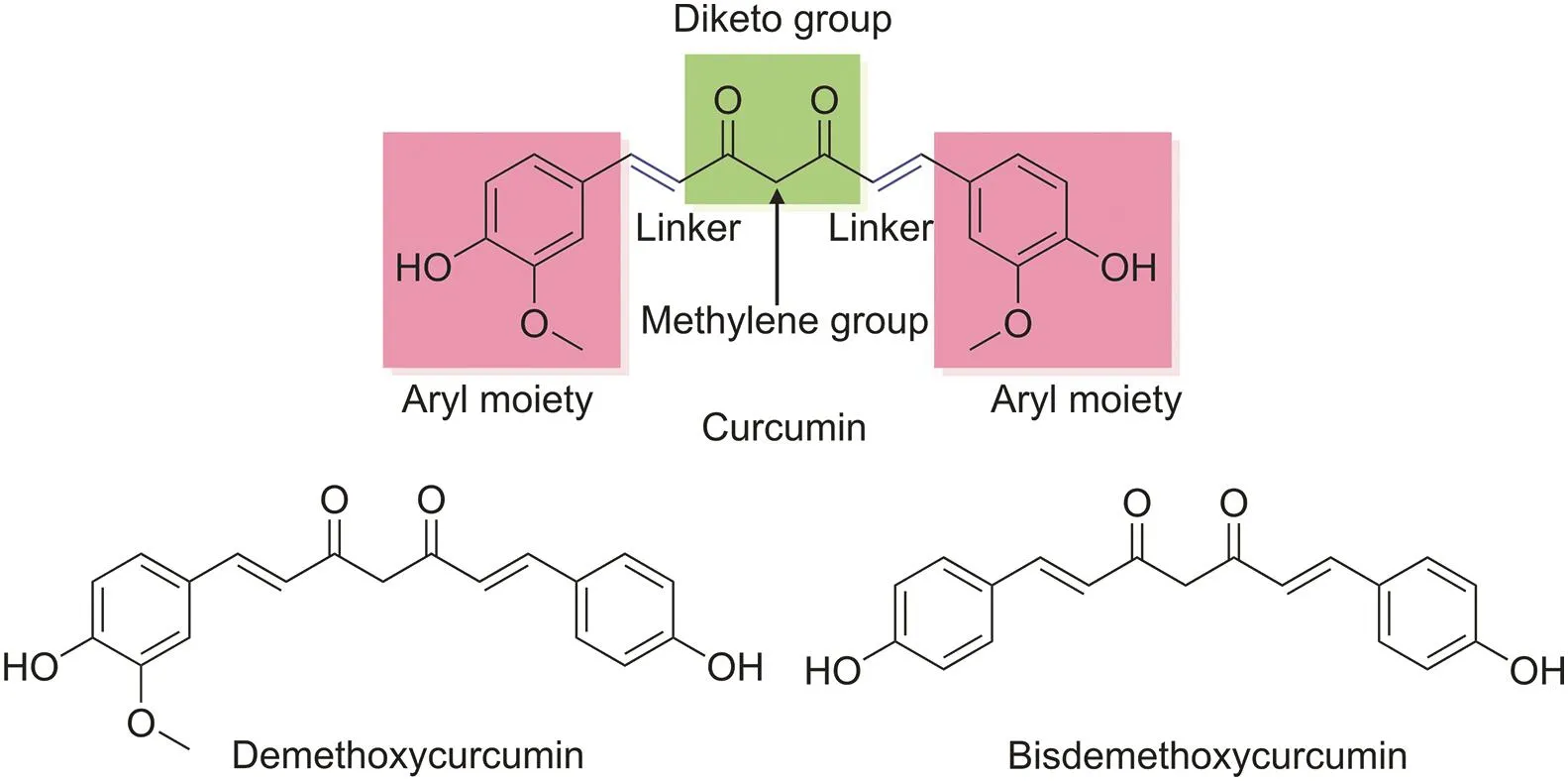
Fig.1.Chemical structures of the main constituents of turmeric.
Additionally,a number of studies have investigated the possible neuroprotective effects of curcumin.Khatri and Juvekar[22]evaluated the effect of curcumin against rotenone-induced cognitive impairment and oxidative and mitochondrial dysfunction in an animal model of neurodegenerative disorder.They found that curcumin enhanced acetylcholine esterase enzyme levels compared to the negative control group,restored motor deficits,and promoted the activities of antioxidant enzymes.Srivastava et al.[23]used arsenic-induced N-methyl-D-aspartate(NMDA)receptor dysfunction to examine the protective efficacy of curcumin in neuronal cells.Curcumin displayed neuroprotective properties by modulating the PI3K/Akt/GSK3βneuronal survival pathway,affecting NMDA receptors and forward signaling through tropomyosin-related kinase B and brain-derived neurotrophic factor in the hippocampus.Furthermore,Yadav et al.[24]demonstrated that simultaneous treatment with curcumin(100 mg/kg body weight)within 28 days improved learning and memory performance in arsenic-induced cholinergic dysfunction in rats.
Numerous curcumin analogs possess different biological activities and are present in diverse biological organisms.These analogs share common structural features with curcumin,which are probably responsible for their comparable pharmacological properties.The essential biological activities of some curcumin analogs found in nature are summarized in Table 1[25-44].

Table 1Representative curcumin analogs found in nature and their biological activities.
The pharmacological potency and natural safety of curcumin and its analogs have led to their use in the treatment and prevention of numerous diseases[45].However,the potential use of curcumin is limited by its inadequate aqueous solubility,low stability in an aqueous environment,poor bioavailability,and inferior cellular uptake[14].Despite enormous efforts to enhance the physicochemical and pharmacological properties of curcumin,medicinal chemistry approaches have failed to improve its potencysignificantly,and no curcumin analogs have been found to be more potent than curcumin.Therefore,further studies are needed to overcome multiple issues associated with the development of curcumin and its derivatives[46].
2.2.Flavonoids
Flavonoids are secondary plant metabolites synthesized in response to stressful conditions and are commonly found in vegetables,seeds,fruits,and beverages.They play an important role against parasites,including pathogens and insects.Structurally,flavonoids are hydroxylated compounds synthesized through the phenylpropanoid pathway and are divided into different classes(e.g.,flavanols,flavanones,flavones,flavanols,anthocyanins,and chalcones)[47].Because of their beneficial pharmacological effects,flavonoids are among the most investigated naturally occurring polyphenolic molecules for treating various diseases[48].The diverse chemical structures of the main flavonoids found in nature,such as quercetin,naringenin,genistein,taxifolin,rutin,and luteolin,are displayed in Fig.2.
Quercetin is the most abundant dietary flavonoid ubiquitously present in various edible plants and plant-derived beverages,such as berries,grapes,apples,citrus,cocoa,onion,tea,and red wine[49].Quercetin has a variety of biological activities,especially relating to its antioxidant properties[50].In a study by Borghi et al.[49],quercetin inhibited titanium dioxide-induced chronic arthritis in mice.Veith et al.[51]demonstrated that quercetin reduced the lipopolysaccharide (LPS)-induced production of the proinflammatory cytokines interleukin-8 and tumor necrosis factor alpha in both patients with idiopathic pulmonary fibrosis and healthy subjects.Furthermore,quercetin displayed anticancer properties by blocking the Akt/mTOR/c-Myc signaling pathway to suppress ribosomal protein RPS19-activated epithelial-mesenchymal transition signaling and ablated the expression of ribosomal proteins[52].By acting as an antiinflammatory and antioxidant agent,quercetin attenuates the severity of cerulein-induced acute pancreatitis[53].Additionally,quercetin exhibits inhibitory activities both in vitro and in vivo against LPS-induced nitric oxide production[54].
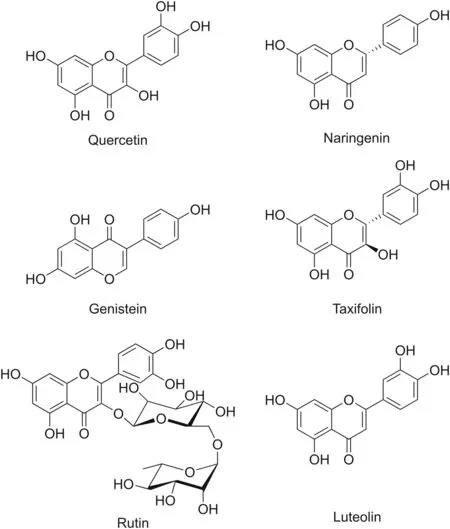
Fig.2.Structures of the various flavonoid compounds.
Rutin,a natural flavone derivative that exists in various natural sources,is a polyphenolic molecule that has antioxidative effects on oxidizing species,such as OH radicals,superoxide radicals,and peroxyl radicals.Moreover,rutin has been widely studied over the past few decades in several human diseases,including inflammation,allergies,tumors,diabetes,hypertension,hypercholesterolemia,and infection.An in vitro study on human fibroblasts(CCD-1112Sk)revealed that rutin displayed numerous antioxidant properties,including the amelioration of the ultraviolet(UV)-induced pro-inflammatory response and reactive oxygen species(ROS)generation,increased the levels of antioxidant agents in living cells(such as superoxide dismutase,glutathione peroxidase,vitamin E,glutathione,and thioredoxin),regulated UV-induced nuclear factor E2-related factor 2 expression[55,56],and inhibited benzo[a]pyrene-7,8-diol-9,10-epoxide-induced cyclooxygenase-2expression[57].
The potential properties of naringenin(a flavonoid belonging to the subclass flavanone)have been investigated in vitro and in vivo.For example,an in vivo study revealed that naringenin could attenuate lead-induced biochemical alterations in the serum,liver,and kidney tissues in animal models.This finding suggests that naringeninplays a protective role against oxidative damage[58].By selectively modulating drug pathways,naringenin enhanced the antitumor activity of the chemotherapeutic agent doxorubicin in human lung adenocarcinoma(A549)and breast cancer(MCF-7)cell lines[59].It was further suggested that naringenin might play an important role in treating human fibrosis[60],inhibiting two-pore channel 2-mediated signaling in angiogenesis[61],protecting neurons againstβ-amyloid toxicity through the regulation of amyloidogenesis and tau phosphorylation in Alzheimer's disease(AD)[62],preventing lipid peroxidation,and inhibiting hepatic cell damage[63].
Similar to other flavonoid compounds,genistein possesses anticancer activity.Genistein,the main isoflavone constituent of soybean,has demonstrated various effects on human health.Rusin et al.[64]investigated the antiproliferative properties of genistein derivatives(modified at the C7 position by various mono-and diunsaturated sugars)in a human colon cancer cell line(HCT116).In preliminary screening for cancer cell proliferation inhibition,the synthesized compounds appeared more potent than the parent compound(genistein).Similarly,the antiproliferative potential of genistein derivatives has been reported in other studies[65-68].For instance,genistein induced cell death and inhibited androgensensitive human prostate adenocarcinoma cell growth through the modulation of some genes involved in carcinogenesis and immunity[69].
Taxifolin and luteolin are flavonoids structurally similar to quercetin.They exhibit a broad range of pharmacological activities.Taxifolin has strong antioxidant properties(e.g.,ROS scavenging),inhibits the synthesis of triglycerides,protects against cerebral ischemic injury,exhibits antiproliferative activity,promotes the apoptosis of numerous tumor cells,acts as a potential P-gp modulator[70],and inhibits amyloid fibrillation and its related toxicity[71].Emerging evidence has revealed that luteolin,a polyphenolic compound that belongs to the flavone subclass,possesses various biological and chemical activities,including protective effects,in a multitude of cardiovascular diseases(e.g.,coronary artery disease,heart failure,and atherosclerosis)[72],inhibits anoctamin 1 chloride channel activity in prostate cancer[73],exerts synergistic antitumor effects on colorectal cancer cells(in combination with oncolytic adenovirus(CD55-TRAIL))[74],and inhibits metastasis in prostate cancer[75].
2.3.Naturally occurring peptides from marine sources
In recent years,naturally occurring peptides(e.g.,hormones,growth factors,neurotransmitters,and anti-infective peptides)isolated from plants and animals have been found to play a vital role in human health.Approximately 7000 peptides/peptidomimetics have been identified in nature[76].Due to the vast number of bioactive peptides available in nature,only those from marine sources are described in this review.As an important bioactive natural constituent,marine peptides are present in many species,such as sponges,seaweeds,ascidians,mollusks,and marine microorganisms[77].Interestingly,because of the harsh environmental conditions(i.e.,very exigent,competitive,and aggressive surroundings)in which they inhabit,marine peptides possess significantly modified structures either in their backbone or side chain compared to peptides of human origin,resulting in potent active molecules.Thus,marine peptides serve as valuable candidates for drug discovery,offering good stability under thermal conditions,higher in vivo bioavailability,and better resistance to enzymatic degradation[77,78].These properties make them superior and more useful in nuclear medicine than terrestrial peptides.
The importance of natural peptides as sources of novel bioactive substances is rapidly increasing.Marine peptides exhibit a variety of biological properties,including anti-infective,anticancer,anticoagulant,antioxidant,and immunomodulatory effects.Over the past few decades,the bioactivity of marine peptides has been studied in both preclinical and clinical settings.In particular,studies investigating the anti-infective effects of marine peptides have attracted significant interest among researchers and clinicians worldwide.In 2017,Ma et al.[79]isolated four novel marine peptides from the gorgonian-derived fungus Aspergillus sp.SCSIO 41501(one cyclic pentapeptide and three linear peptides).Among these peptides,they revealed two molecules,namely,aspergillipeptide D and aspergillipeptide E(Fig.3),that exhibit antiviral activity against herpes simplex virus type 1 with IC50values of 9.5 and 19.8μM,respectively.
In addition,peptides from marine environments have shown antimicrobial activities against Staphylococcus aureus,Klebsiella pneumoniae(proline-rich peptides isolated from the hemolymph of Rapana venosa snails)[80],E.coli(arenicin-1,isolated from Polychaeta Arenicola marina)[81],Acinetobacter baumannii,Pseudomonas aeruginosa(P.aeruginosa),B.subtilis(misticalins,isolated from marine mussels(Mytilus spp.))[82],Vibrio cholerae,Proteus mirabilis,Salmonella paratyphi,Streptococcus pyogenes,and Klebsiella oxytoca(dromidin,isolated from the marine crab Dromia dehaani)[83].
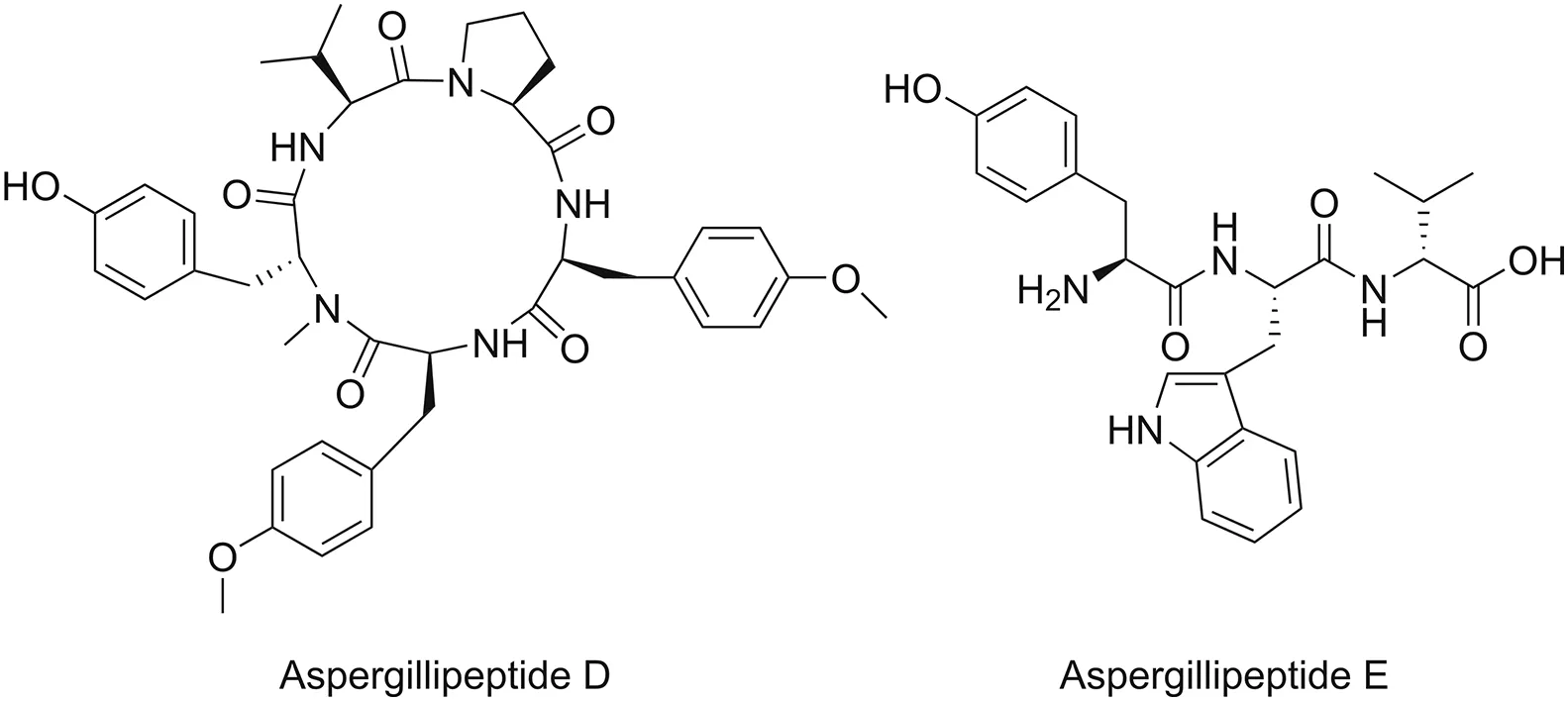
Fig.3.Chemical structures of aspergillipeptide D and aspergillipeptide E.
Marine peptides can significantly reduce blood pressure by modulating angiotensin I-converting enzyme(ACE).ACE is a zinc metalloprotease originally isolated from horse blood and comprises two zinc entities,active catalytic sites,and a dipeptidyl carboxypeptidase.This enzyme plays an essential role in regulating blood pressure as it stimulates the conversion of angiotensin I to the potent vasoconstrictor angiotensin II and catalyzes the degradation of the vasodilator bradykinin[84,85].Ko et al.[86]examined the ACE inhibitory activity of a purified peptide from an enzymatic hydrolysate of Chlorella ellipsoidea and found that the peptide displayed promising ACE inhibitory activity with an IC50value of 128.4μM.They also found that the peptide significantly decreased systolic blood pressure in spontaneously hypertensive rats.These results highlighted the role of ACE as a functional therapeutic approach in the treatment of hypertension.
Many marine-derived peptides have anticancer activity against cancer cell lines,and a few of them have been evaluated in animal models.Using chemical synthesis,Anand et al.[87]studied the biological activity of euryjanicin A,a proline-containing cyclic heptapeptide,which was previously isolated from the marine sponge Prosuberites laughlini.They found that the peptide showed potent anticancer activity against the human colon cancer cell line HeLa.Furthermore,Tran et al.[88]investigated the anticancer potential of marine peptides.They demonstrated that 12 isolated peptides from the Australian marine sponge Pipestela candelabra could selectively inhibit the growth of human prostate cancer cells(PC3),with IC50values ranging from picomolar(48.4 pM)to submicromolar(2.87μM).
Other naturally occurring peptides from marine environments as well as their related pharmacological activities and chemical structures that have been successfully elucidated using spectroscopic techniques are presented in Fig.4 and Table 2[89-93].
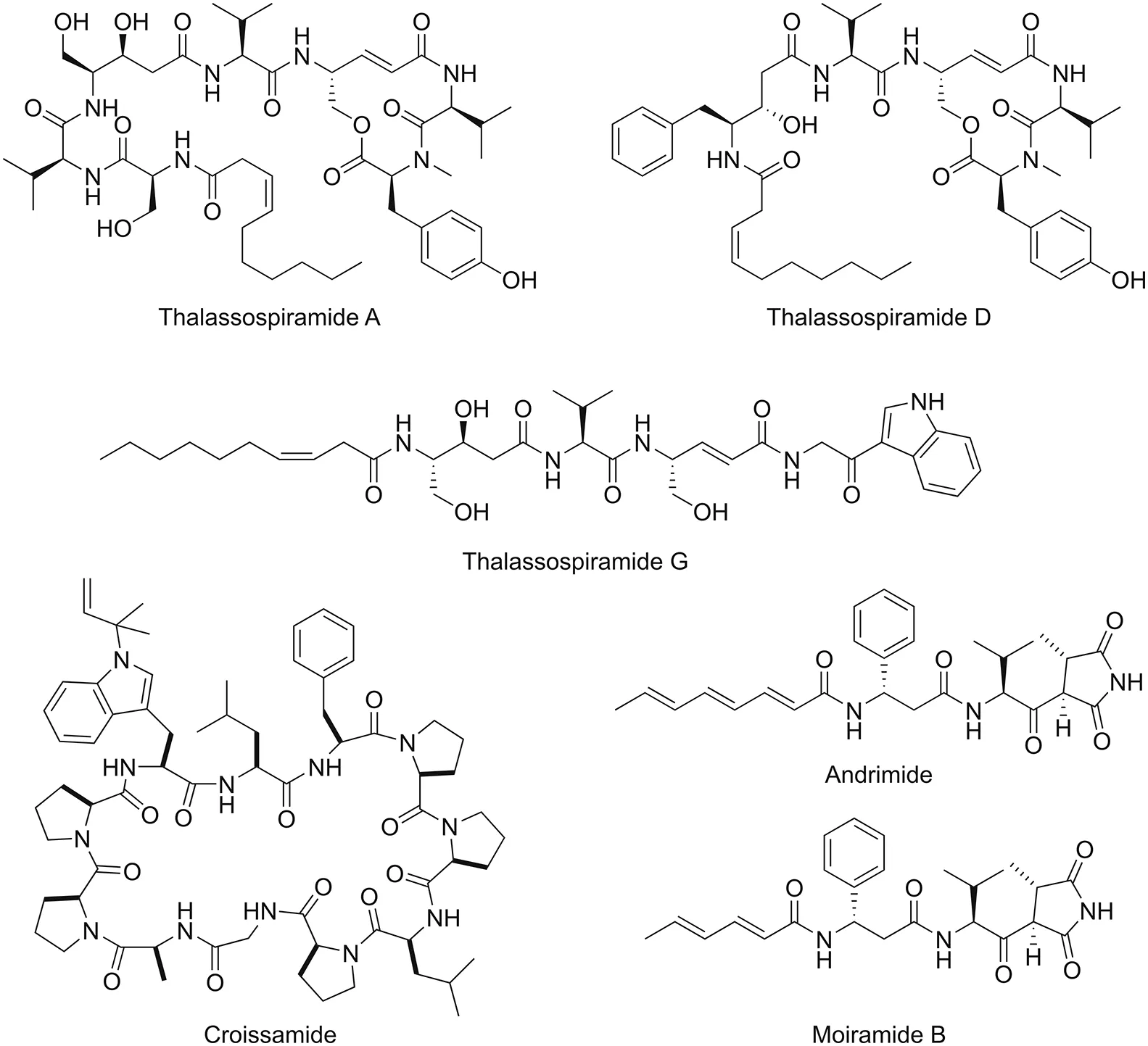
Fig.4.Chemical structures of naturally occurring peptides from marine environments.
3.Radiolabeling of nature-derived products
A radiopharmaceutical,also known as a radiolabeled compound,is a unique pharmaceutical formulation composed of a radioactive substance attached to a bioactive molecule intended for use in the diagnosis or treatment of diseases.In nuclear medicine,radiopharmaceuticals can be administered orally,by inhalation or via injection.The energy emitted can be detected by non-invasive imaging modalities using PET or single-photon emission computed tomography(SPECT),thus providing functional information about the target tissue ororgan.This technique relies on the selective binding of a radiopharmaceutical agent to a specific tissue or organ system in a selective manner[94-96].
The development of new radiopharmaceuticals in nuclear medicine,either for diagnostic or therapeutic purposes,comprises a number of steps.There is no general rule,but at least seven major steps are required to generate natural product-based radiopharmaceuticals(Fig.5A).In contrast,the production of radiopharmaceuticals using chemical synthesis requires only five steps(Fig.5B).
For natural product-based radiopharmaceuticals,production generally starts with the screening or exploration of natural product sources in nature and searching for a lead compound with relevance to human diseases,followed by the isolation of the compounds of interest.These two initial steps are essential and usually require tremendous effort,time,and resources.Once the isolated products have been obtained,their molecular structures are elucidated,commonly by nuclear magnetic resonance spectroscopy,mass spectrometry,infrared spectroscopy,X-ray crystallography,and other spectroscopic methods[97,98];the products are subsequently subjected to biological studies(e.g.,anticancer,anti-infective, antioxidant, and anti-inflammatory assays).Depending on the complexity of the molecules and their potential molecular targets(related to biological activities),methods for radiolabeling the chosen compounds are developed.Indeed,the development of natural product-based radiopharmaceuticals requires more work than that of radiopharmaceuticals of synthetic origin.As a result,the number of natural product-based radiopharmaceutical studied in recent years is considerably low.
Ideally,the introduction of a radionuclide into a bioactive compound should not affect the active sites of the molecule to produce a radiopharmaceutical with an activity profile comparable to that of the parent structure[8].In reality,this process is challenging and often requires the molecular modification of lead compounds(precursors)to enable radionuclide installation.Nevertheless,functionalized radiopharmaceuticals can suffer from a dramatic decrease in the desired bioactivity as a result of adding or changing specific groups[9].Moreover,the radiolabeling strategies developed often include reaction optimization to produce radiolabeled compounds with high radiochemical yields in a short reaction time[99,100].Several widely used radioactive isotopes and their applications in nuclear medicine are summarized in Table 3[101-104].

Table 2Peptides isolated from marine environments and their biological activities.
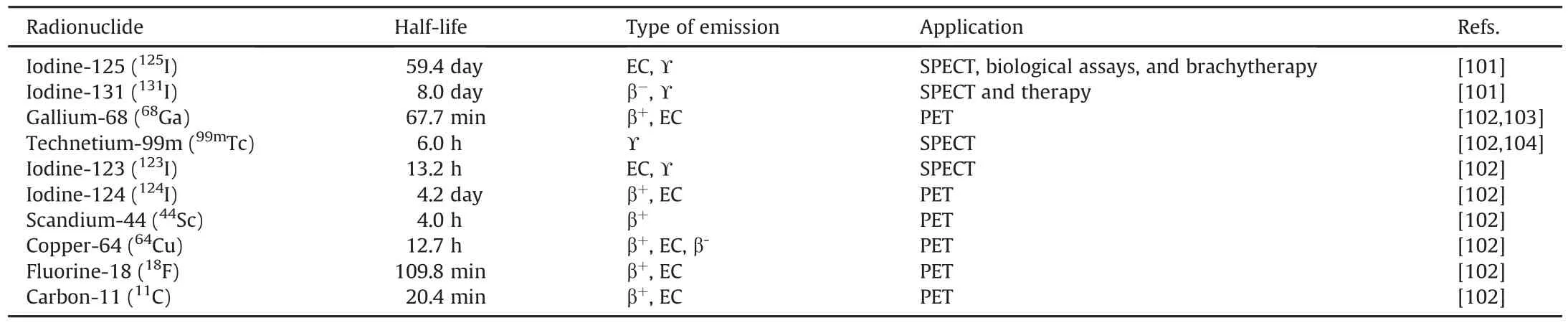
Table 3The characteristics and applications of medically relevant radionuclides currently employed in nuclear medicine.
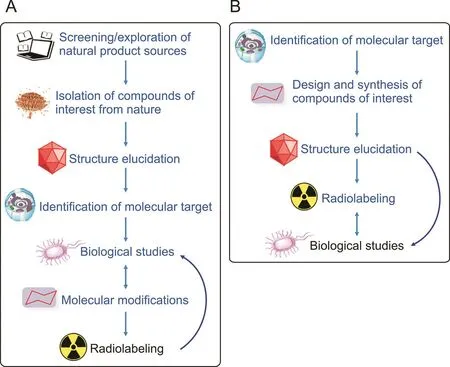
Fig.5.Main steps in the synthesis of(A)natural product-based radiopharmaceuticals and(B)radiopharmaceuticals of synthetic origin.
Until recently,there have been very few reports on the synthesis of radiopharmaceuticals using natural chemical templates.Although the utilization of natural products as a template for the development of new radiopharmaceuticals has been limited,some natural product-based radiopharmaceuticals have been developed in recent years.One example is technetium-99m-labeled triazole-Lys-bombesin,which was synthesized using the click chemistry approach and demonstrated specific recognition of gastrinreleasing peptide receptors(GRPRs)in MCF-7 cells[105](Fig.6).Bombesin,a peptide that was first isolated from the skin of frogs in 1970,is a tetradecapeptide analog of human GRPRs that has GRPR-binding ability.Due to their favorable activity targeting GRPRs,several bombesin analogs and their corresponding radiolabeled molecules have been developed[106-108].
3.1.Radiolabeling strategies for curcumin and its analogs
Radiolabeled curcumin and its analogs with radiometals(e.g.,gallium(Ga)-68)have been investigated in some pharmacological studies because of their potential as therapeutic agents in various human diseases.Asti et al.[109]reported the synthesis of Ga-68-labeled curcumin,along with two other analogs,namely,diacetylcurcumin and bis(dehydroxy)curcumin,for the visualization of βamyloid plaques in AD(Fig.7).All Ga-curcuminoid complexes showed a high binding affinity for synthetic β-amyloid filaments and demonstrated high internalization in the lung cancer cell line(A549).These complexes were obtained after 10 min of reaction with high purity and high radiochemical yields and showed in vitro stability in sodium chloride solution and human serum in transchelation and transmetalation experiments.Similarly,using the same Ga-curcuminoid complexes,Rubagotti et al.[110]studied βamyloid plaque formation in Tg2576 mice.Post-mortem brain cryosections of Tg2576 showed that allnat/68Ga-curcuminoid complexes failed to detect β-amyloid pathology in vivo,possibly because of the poor stability of the complexes in vivo.However,the complexes maintained a high affinity for β-amyloid fibrils and plaques in vitro.
The design of68Ga-based radiopharmaceuticals may also involve utilizing a bifunctional chelator(BFC)agent capable of coordinating the metal bound to a chelator,called an indirect labeling procedure.Moreover,organic spacers can be exploited to expand the distance between the bioactive molecules and BFCs to prevent steric impact,resulting in more flexible complexes that can more easily bind to their desired molecular targets[111].Orteca et al.[103]reported the synthesis of two68Ga-labeled compounds based on acurcumin scaffold,namely(1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione-1,4,7-triazacyclononane-1-glutaric acid-4,7-diacetic acid(NODAGA-C21)and (1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione)-1,4-bis(carboxymethyl)-6-[bis(carboxymethyl)]amino-6-methylperhydro-1,4-diazepine(AAZTA-PC21),in a four-step procedure(Fig.8)[103].Using the BFC approach,curcumin analogs were connected to the NODAGA and AAZTA chelators.The conversion of the keto-enol group in curcumin into a pyrazole ring in AAZTA-PC21 significantly improved the stability of the complexes.
Furthermore,Rokka et al.[112]reported the synthesis of[18F]-curcumin derivatives to investigate β-amyloid in AD(Fig.9).The radiolabeled compound was obtained in a considerably good radiochemical yield from a one-pot synthesis procedure through the nucleophilic[18F]-fluorination of 2-[2-(2-azidoethoxy)ethoxy]ethyl tosylate,followed by a click reaction with alkyne-containing curcumin.Although the radiotracer exhibited fast blood clearance,it appeared to have a poor ability to cross the blood-brain barrier,possibly due to its unsuitable molecular weight and the presence of a bulky fluoropegylated tail attached to the triazole.
Additionally,curcumin can be directly labeled with radioiodine(e.g.,125I)using iodogen-coated glass tubes as an oxidizing agent,as demonstrated by Samuel and co-workers[113].Radiolabeled curcumin was obtained with a radiochemical yield of~75% and a maximum radiochemical purity of 95% after purification by extraction using chloroform.In this study,the structure of125I-curcumin was not determined;however,the possible structure is shown in Fig.10.
Organic molecules bearing radionuclide iodine significantly contribute to the field of nuclear medicine,especially nuclear molecular imaging.Different methods for the radioiodination of small molecules and their outcomes have been reviewed by Dubost et al.[114].In this comprehensive review,radioiodination methodsincluded direct electrophilic aromatic substitution,isotopic and halogen exchange,iododestannylation,iododesilylation,iododeboronation,and transition-metal-mediated halogen approaches,which can be adopted for the synthesis of natural product-based radiopharmaceuticals.Among these methods,direct electrophilic aromatic substitution may be more suitable for the radioiodination of natural products,typically for amine-and phenol-containing compounds.The addition of one or more iodine atoms to the bioactive molecule insignificantly alters the structure of the parent molecule,thereby maintaining biological activity[115].Molchanova et al.[116]investigated the effect of halogen atoms(iodine,fluorine,bromine,and chlorine)on the phenyl ring of peptoids and found that halogenation had no effect on the activity against E.coli and P.aeruginosa.
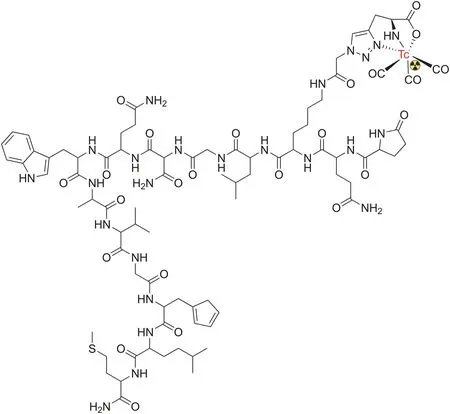
Fig.6.Structure of99mTc(CO)3-triazole-Lys3-bombesin.
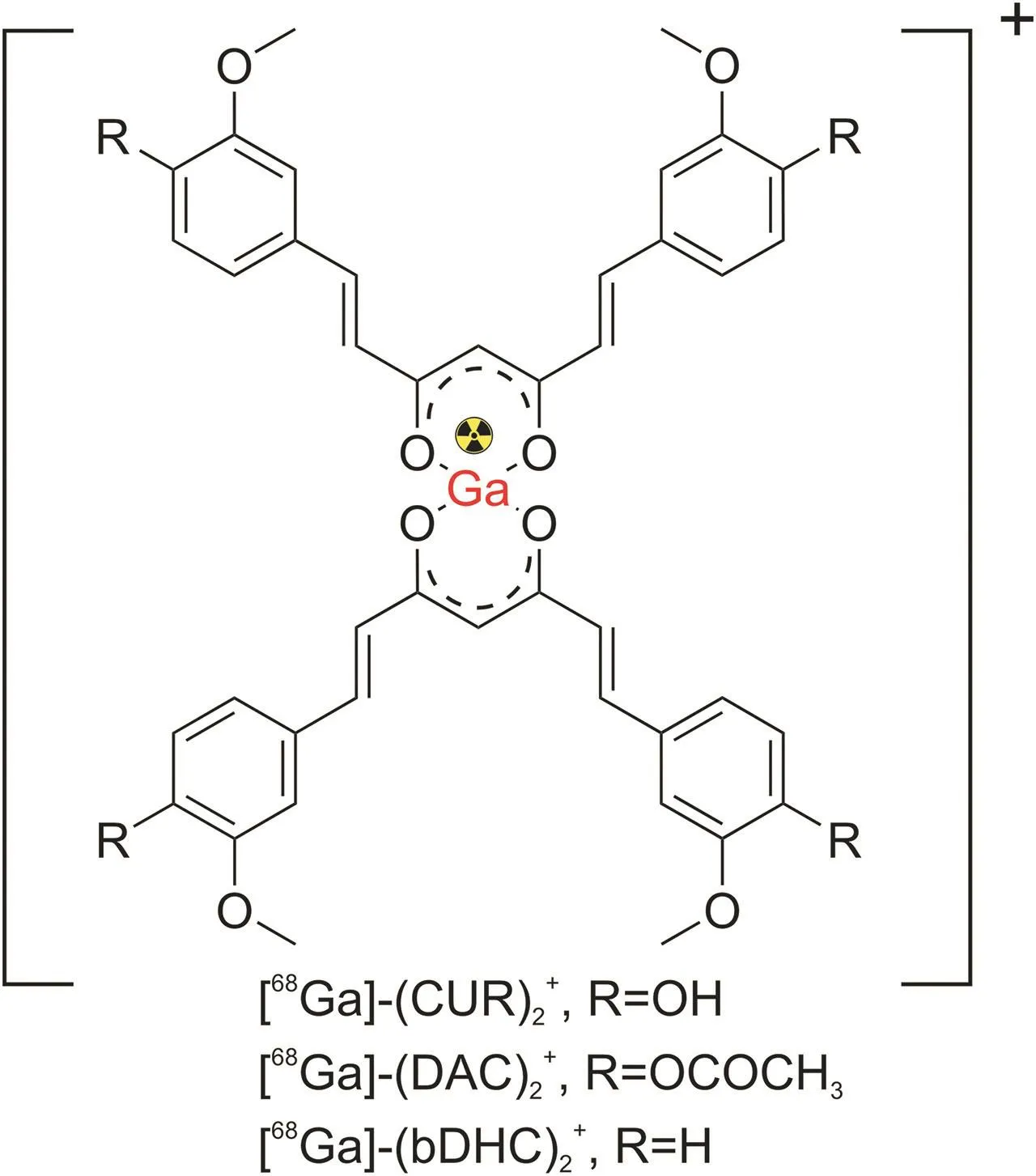
Fig.7.[68Ga]-radiolabeled curcumin and its analogs.The counter ion is generally chloride but usually depends on the medium.
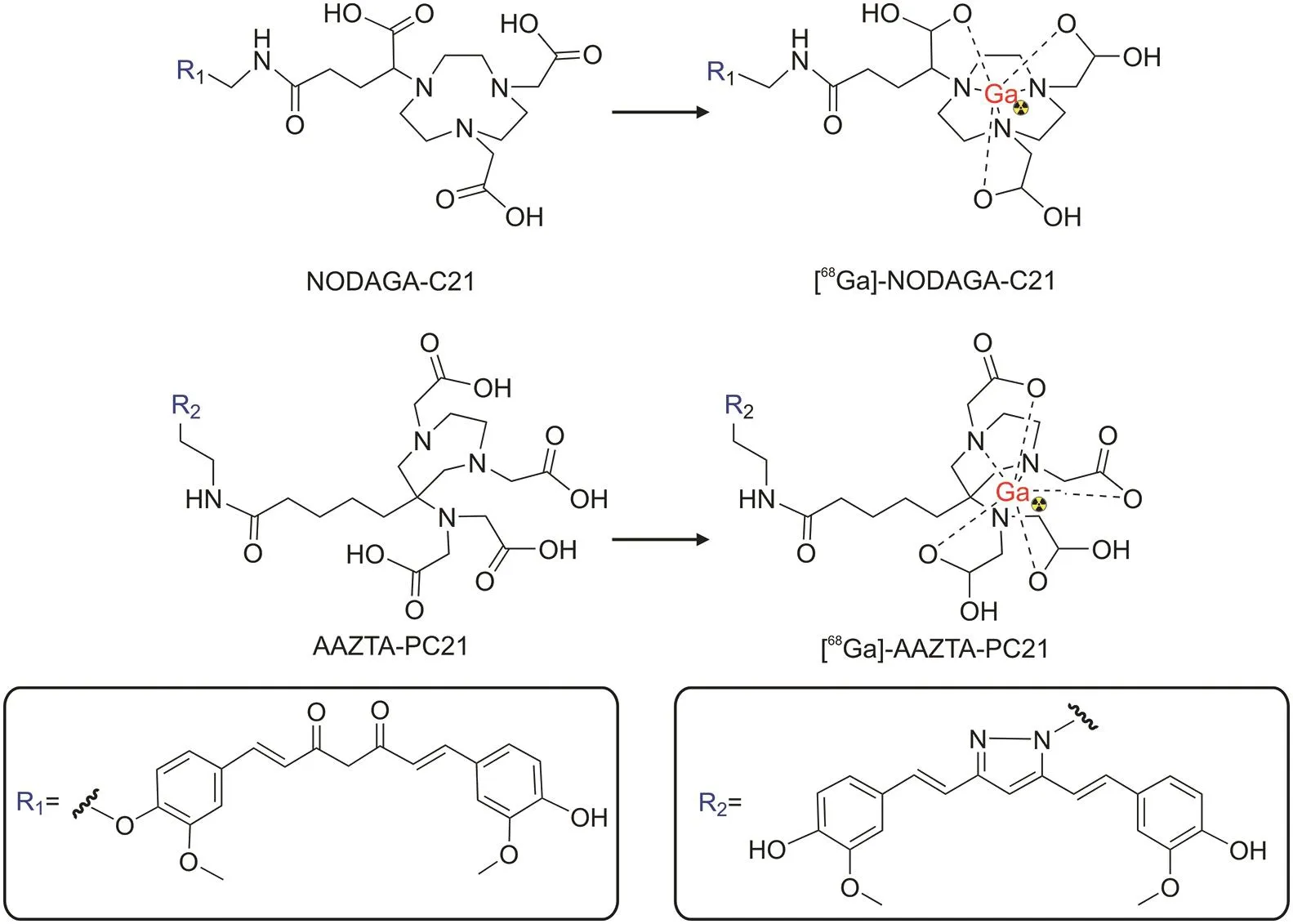
Fig.8.Radiolabeling of NODAGA-C21 and AAZTA-PC21 with gallium-68.(Reprint from Ref.[103]with permission).
The presence of electron-donating groups,such as amines and phenols,in the aryl moiety provides rapid iodination by activating the carbons on the ring to undergo electrophilic aromatic substitution.Accordingly,molecules containing phenols,aniline derivatives,and alkyl aniline functionalities are more likely to be radioiodinated[115,117].Although they have not previously been reported,diverse curcumin analogs,including demethoxycurcumin and bisdemethoxycurcumin,isoeugenol,caffeic acid,ferulic acid,and dehydrozingerone,could potentially be labeled with radioiodine by direct electrophilic aromatic substitution.With phenol,the substitution of the iodine atom might occur at the ortho position of the hydroxyl group(Fig.11).
3.2.Radiolabeling strategies for flavonoid compounds
Most flavonoids found in nature are phenol rich.This property makes them suitable as promising precursors for the synthesis of labeled compounds,especially using direct iodination methods as previously discussed in Section 3.1 or other iodination methods,such as iododestannylation.Moreover,some flavonoids have been labeled with other radionuclides,such as fluorine-18,technetium-99m,and scandium-46.
Fuchigami et al.[118,119]developed radiolabeled-related flavonoid molecules, including flavones, aurones, styrylchromones,and chalcones,as PET and SPECT imaging probes for amyloid deposits in the brain.They found that a styrylchromone derivative,[123I]-SC-OMe,showed higher in vitro binding affinity(Ki=20.8 ± 0.69 nM)than other derivatives and successfully recognized the positive prion deposits in the prion disease mouse model.The radiosynthesis of these compounds was accomplished by iododestannylation from the corresponding tributyltin precursors.For instance,radiolabeled aurone compounds were prepared in radiochemical yields of 48.2%-67.6%.Briefly,the tributyltin compounds were converted into their corresponding bromo compounds under Pd(0)-catalyzed reaction conditions,followed by iododestannylation in chloroform at room temperature to generate the radiolabeled aurone derivatives(Fig.12)[120].
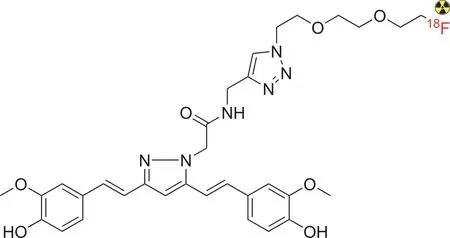
Fig.9.Structure of the[18F]-curcumin derivative.
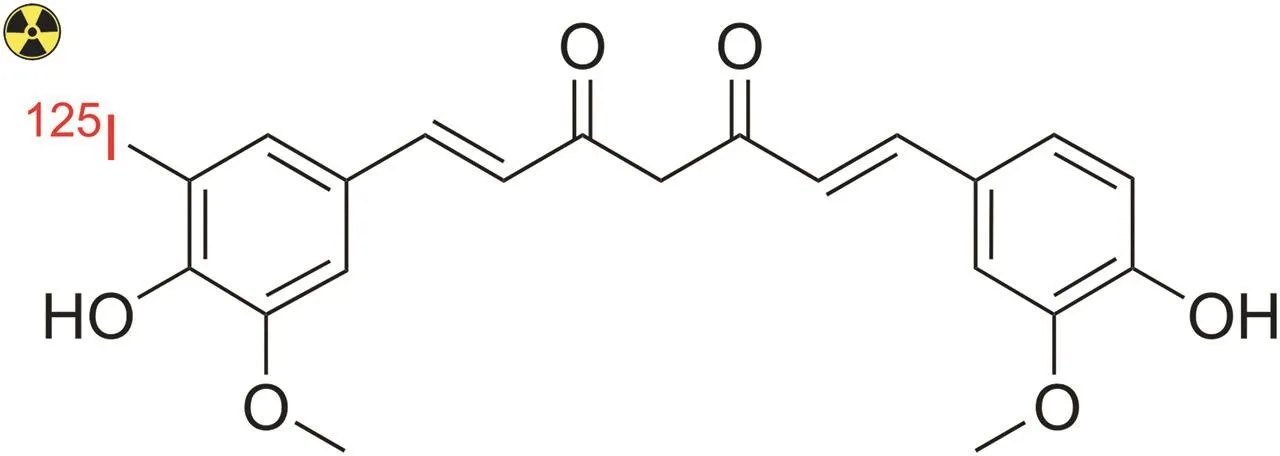
Fig.10.Predicted chemical structure of mono-iodinated[125I]-curcumin.
Additionally,some novel chalcone derivatives have been incorporated with technetium-99m by the chelation ligands monoamine-monoamide dithiol(MAMA)and bis-amino-bis-thiol(BAT)for imaging Aβ plaques(Fig.13)[121].The compounds were synthesized via a simple ligand exchange reaction utilizing the precursor99mTc-glucoheptonate in 46%-95% radiochemical yields and a radiochemical purity greater than 95% after purification by HPLC.An in vitro binding assay showed that the length of the carbon linker(n)between technetium-99m complexes and the chalcone moiety played an essential role in binding to the Aβ(1-42)aggregates.Compounds with shorter carbon linkers exhibited greater binding.However,the different ligands(MAMA and BAT)did not alter the binding affinity[121].
Recently,Allott et al.[122]reported the synthesis of a fluorine-18-labeled chalcone derivative based on a CDg4 scaffold as a dualmodality imaging agent(PET and fluorescence)for detecting glycogen in cancer tissues(Fig.14).The compound was realized by a simple SN2 displacement of the tosyl group with[18F]fluoride in a 5.1%±0.9% radiochemical yield and excellent radiochemical purity(>98%).In vitro fluorescence imaging indicated that the compound selectively accumulated in the glycogen-containing cells.PET imaging in normal mice and metabolite analysis suggested favorable hepatobiliary intestinal elimination and fast metabolism of the radiolabeled chalcone.
Additional radiolabeled flavonoids,such as quercetin,rutin,hesperitin,and apigenin,and their radiolabeling strategies have been documented(Table 4)[115,123-128].For instance,Hosseinimehr et al.[123]labeled quercetin with iodine-125 and found that the labeled compound exhibited a potential role in inducing DNA damage and thus could be considered a targeted radiotherapy agent.Similarly,successful radiolabeling of quercetin with iodine-131 was demonstrated by Sriyani et al.[124].Furthermore,radioiodination has been performed on rutin,hesperetin,and apigenin[126-128].
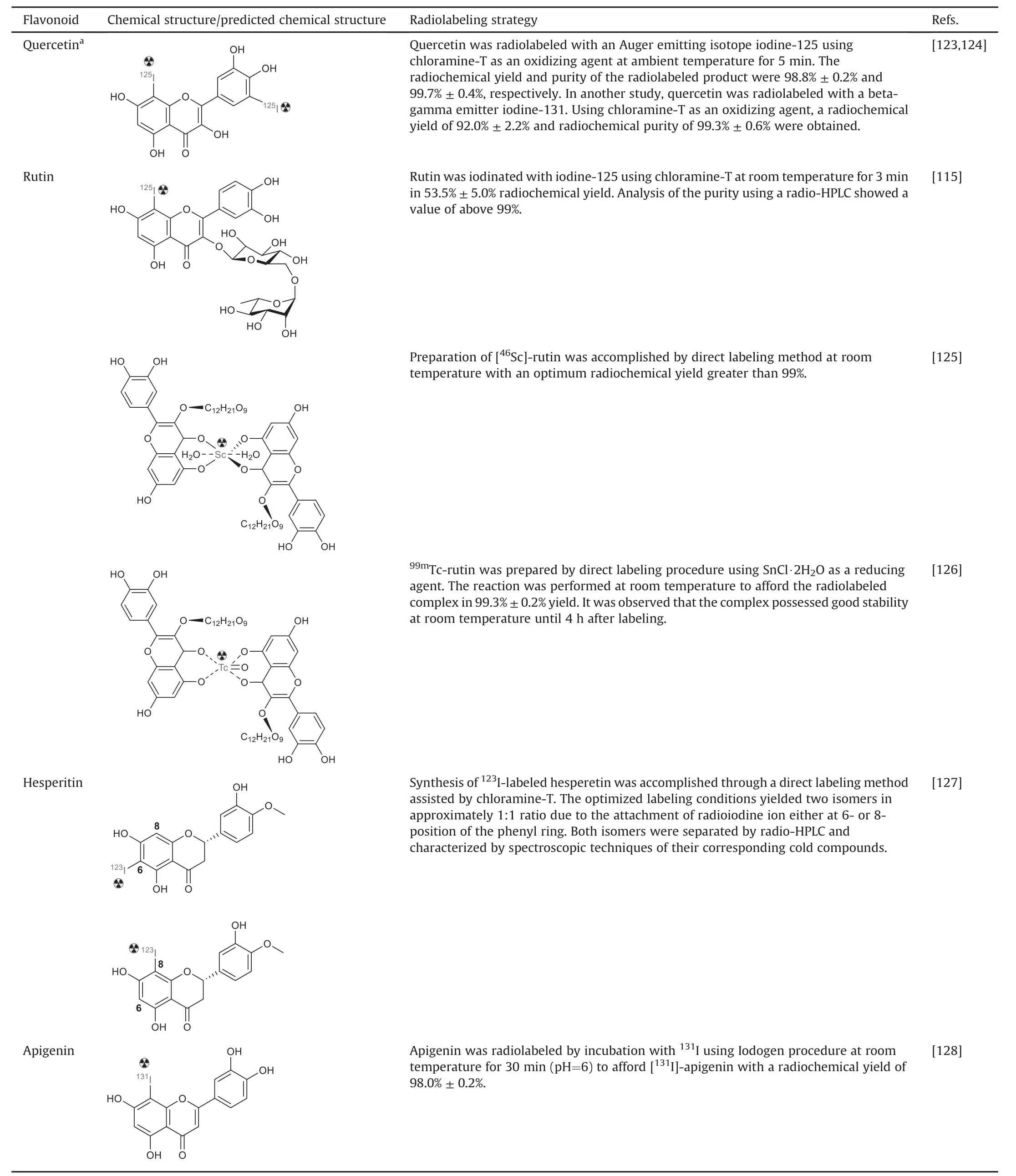
Table 4Reported radiolabeled flavonoids and radiolabeling strategies.

Fig.11.Predicted outcomes of radioiodinated curcumin analogs.Only monoradioiodinated products are shown.In reality,multiple iodinated products in one molecule might occur depending on the compound's chemical nature.
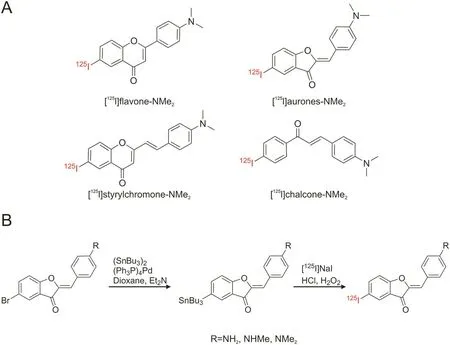
Fig.12.(A)The chemical structures of radiolabeled flavonoids as imaging agent candidates for prion deposits and Aβ plaques and(B)radiosynthesis of flavonoid derivatives(aurones)via iododestannylation.(Reprint from Ref.[120]with permission).
3.3.Radiolabeling strategies for marine peptides
Due to their diverse biological activities,peptides have been of interest for many decades.Additionally,diverse radiolabeling options are available for peptides via radioactive halogen isotopes(e.g.,fluorine-18,bromine-76,iodine,and astatine-211)and radiometals(e.g.,copper-64,gallium-68,zirconium-89,actinium-225,yttrium-90,and lutetium-177).Hence,a number of peptides have been developed as radiolabeled probes for use in nuclear medicine for diagnosis or therapy,with some becoming the primary treatment options for the management of cancers[129,130].Radiolabeled peptides possess pharmacokinetic properties comparable to those of small molecules,including a high degree of binding selectivity for their receptor and fast clearance from the body via secretory organs,such as the kidneys.Furthermore,peptides are generally non-immunogenic[131,132].
In addition to radiolabeled bombesin,some peptide-based radiopharmaceuticals have been used clinically.For example,[68Ga]Ga-DOTA-peptides([68Ga]Ga-DOTA-TOC,[68Ga]Ga-DOTA-NOC,and[68Ga]Ga-DOTA-TATE),which specifically bind to somatostatin receptors in neuroendocrine tumors,were developed based on a synthetic version of the naturally occurring hormone somatostatin[133].Although radiolabeled peptides have become an important class of molecules for diagnosis and therapy in nuclear medicine,the development of marine peptide-based radiopharmaceuticals has not yet been explored,and examples are rare.

Fig.13.Chalcone derivatives incorporated with technetium-99m through the(A)monoamine-monoamide dithiol and(B)bis-amino-bis-thiol ligand.(Reprint from Ref.[121]with permission).

Fig.14.18F-labeled chalcone derivative based on the CDg4 scaffold.
Recently,dolastatin 10 was utilized as a potential agent to improve the efficacy of immunoconjugates in cancer therapy.Dolastatin 10 is a peptide originally isolated from the sea hare Dolabella auricularia that exhibits potent cytotoxicity by inhibiting microtubule construction and tubulin-dependent guanosine triphosphate hydrolysis,which results in cell death.Because of their potential cytotoxic properties,several dolastatin 10 derivatives have been synthesized,which led to monomethyl auristatin E(MMAE)and monomethyl auristatin F(MMAF)[134,135](Fig.15).Boswell et al.[136]reported the synthesis of111In-labeled anti-TENB2-MMAE,an antibody-drug conjugate targeting the transmembrane protein with epidermal growth factor-like and two follistatin-like domains in human prostate tumors.Recently,Adumeau et al.[137]radiolabeled trastuzumab immunoconjugates bearing an MMAE toxin and positron-emitting radiometal zirconium-89.The compound exhibited good visualization of tumor tissues in HER2-expressing breast cancer as well as significant antitumor activity.
Several marine peptides, with or without chemical modifications,are compatible with SPECT and PET isotopes.Currently,there are four general strategies for the addition of a radionuclide to a natural peptide,and the choice of each strategy depends on the radionuclide used:pendant labeling,integrated labeling,prosthetic group incorporation,and direct labeling.Radiometals(e.g.,68Ga,111In,and64Cu)are the most commonly used radionuclides in the pendant-labeling approach.This strategy requires BFC to be conjugated to the peptide sequence to form a stable complex[138].Several BFC chelators have been designed,synthesized,and developed,including cyclic chelators(e.g.,1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid, 1,4,7-triazacyclononane-1,4,7-trisacetic acid, 4,10-bis(carboxymethyl)-1,4,7,10-tetraazabicyclo[5.5.2]tetradecane,and 1,4,7,10-tetrakis(carbamoylmethyl)-l,4,7,10-tetraazacyclododecane)and acyclic chelators(e.g.,diethylene triamine pentaacetic acid and 6-hydrazinonicotinic acid)[138,139](Fig.16).Although the thermodynamic stabilities of cyclic and acyclic chelators appear to be very similar,cyclic chelators are generally more kinetically inert than acyclic chelators[139].

Fig.15.The structures of dolastatin 10,monomethyl auristatin E(MMAE),and monomethyl auristatin(MMAF).

Fig.16.Representative cyclic and acyclic bifunctional chelator agents capable of chelating diverse radiometals. DOTA: 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid;NOTA:1,4,7-triazacyclononane-1,4,7-trisacetic acid;CB-DO2A:4,10-bis(carboxymethyl)-1,4,7,10-tetraazabicyclo[5.5.2]tetradecane; TCMC: 1,4,7,10-tetrakis(carbamoylmethyl)-l,4,7,10-tetraazacyclododecane;DTPA:diethylene triamine pentaacetic acid;HYNIC:6-hydrazinonicotinic acid.
Ideally,chelators should possess a high degree of thermodynamic stability and kinetic inertness and generate a low number of isomers during the reaction with a radiometal.Furthermore,the chelators should maintain high hydrophilicity to enhance blood and renal clearance and exhibit good radiation resistance caused by radiolysis[140].The attachment of a radionuclide at a specific site(chelation site),usuallyat the N-or C-terminus far from the binding functionalities,is crucial to minimizing the loss of binding affinity and pharmacological activity of the peptide radiopharmaceutical.Incorporating BFC into the peptide increases biodistribution in vivo and the stability of the radiolabeled products in the human body[111,141].The major disadvantage of using BFCs is their slow radiolabeling kinetics,which often requires high reaction temperatures and longer reaction time that may not be suitable for heat-sensitive peptides[140].
The second strategy for peptide labeling is integrated labeling.As its name suggests,this approach aims to incorporate a radiometal with a peptide structure.In this case,the radioactive isotope is trapped within the target peptide as a result of peptide cyclization[138].Unfortunately,this method is relatively challenging and has rarely been studied.Moreover,for small molecules,integrated labeling might significantly alter biological activity;therefore,it is only suitable for large molecules where the influence of the radiometal size can be minimized[142].Hom and Katzenellenbogen[143]reported the synthesis of a tetradentate oxorhenium(V)ligandtechnetium-99m complex using an integrated labeling approach to visualize estrogen receptors in breast cancer(Fig.17)[143].Although the complex is relatively stable,it exhibits a negligible binding affinity for the estrogen receptor,which emphasizes that small molecules are unsuitable for integrated labeling.
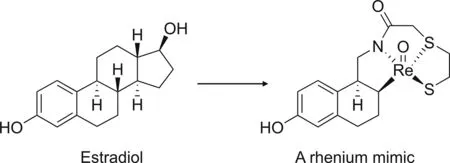
Fig.17.An example of the integrated labeling approach.(Reprint from Ref.[143]with permission).
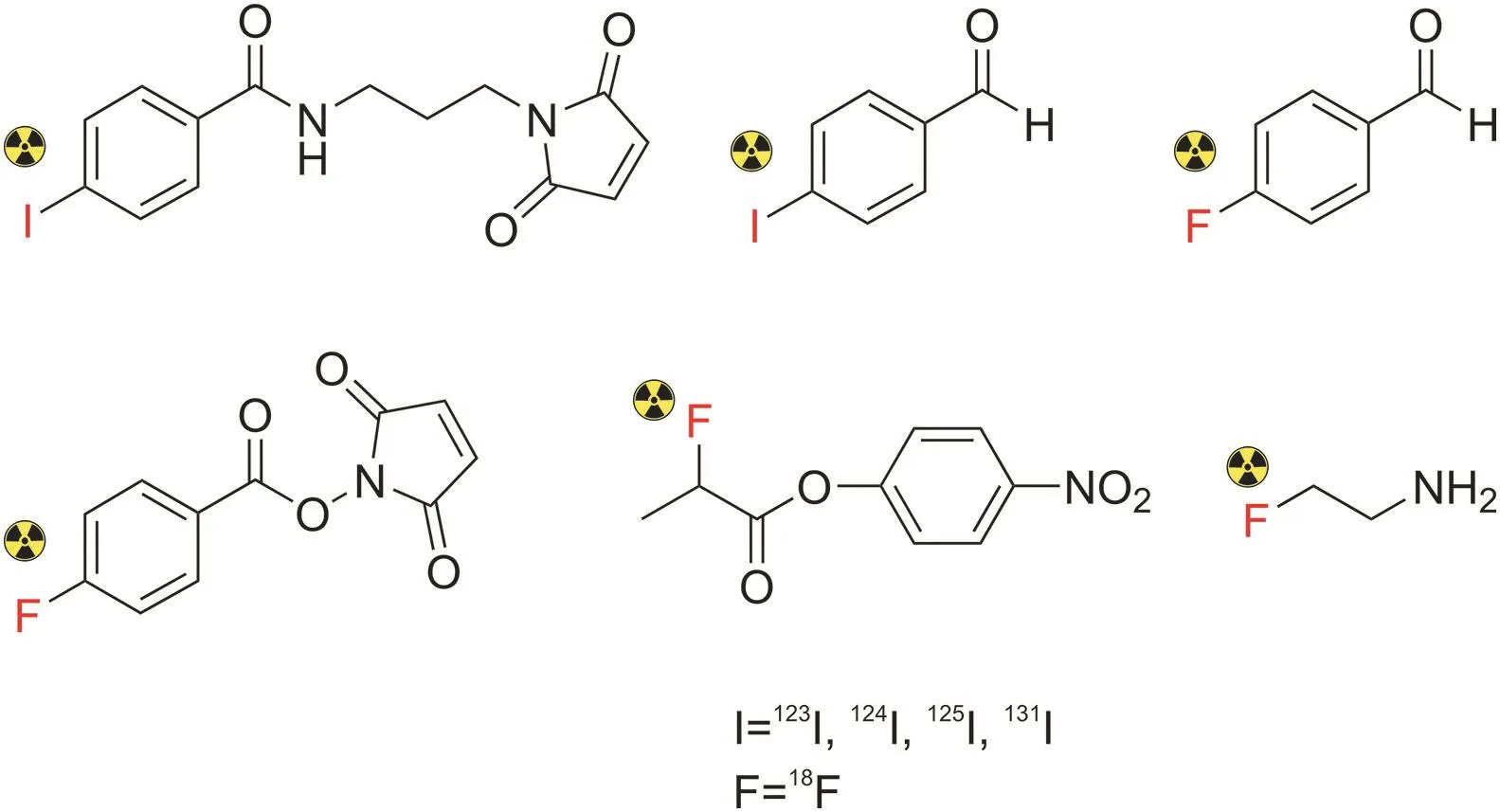
Fig.18.Chemical structures of common prosthetic groups used for the labeling of peptides with radioactive isotopes:iodines and fluorine.
On the contrary,the prosthetic group-labeling strategy is suitable for radionuclides with lower atomic masses,including11C,18F,and radioiodines.In recent years,the development of prosthetic groups has dramatically impacted the field of nuclear medicine[140,144](Fig.18),particularly18F-labeled peptides.In this strategy,small prosthetic groups,such as reactive amine,thiol,and aminoxy moieties,are introduced into the amino side chain of the peptide in one-or two-step procedures,followed by the incorporation of a radionuclide.For example,the incorporation of18F into a peptide can be achieved by nucleophilic aromatic substitution or nucleophilic acyl substitution under mild conditions.The main advantage of this approach is the improvement in the reaction rate and radiochemical yield[138,140].However,this method has several challenges,such as difficulty in achieving site-specific conjugation,high time consumption,and relatively low radiochemical yields[129,132].Moreover,the presence of prosthetic groups could affect the chemical properties of radiolabeled compounds,such as charge or lipophilicity,leading to alterations in biological activities[145].Similarly,Vorobyeva et al.[146]radiolabeled the anti-HER2 DARPin(HE)3-G3 peptide using direct and indirect radioiodination to produce[125I]I-(HE)3-G3 and[125I]I-PIB-(HE)3-G3,respectively.Both radiolabeled peptides showed an equal tumor uptake.However,indirect labeling using N-succinimidyl-para-iodobenzoate showed higher tumor-to-organ and tumor-to-blood ratios than direct labeling.

Fig.19.Examples of the direct radioiodination strategy for marine peptides and their possible outcomes.
The direct labeling approach using iodine and fluorine has become increasingly widespread.This strategy is based on the electrophilic substitution of radionuclides at tyrosine and phenylalanine residues or the direct conjugation of radionuclides to the peptides[111].Direct labeling using radioiodine is carried out by the same mechanism as the iodination of proteins,starting with the generation of electrophilic iodine species using chloramine T or iodogen,which subsequently reacts with the tyrosine residues of the peptide[130].Using this strategy,some marine peptides containing reactive tyrosine or histidine residues could potentially be labeled with radioiodine(Fig.19).
4.Conclusion
Recent advances in the screening,isolation,purification,and characterization of natural products are constantly being refined,allowing the implementation of theories for the rapid and efficient development of new radiopharmaceuticals derived from nature.However,knowledge regarding the contribution of natural products to the development of radiopharmaceuticals is currently limited.In this review,three compound classes(curcumin and its analogs,flavonoids,and marine peptides)and their potential as favorable radiopharmaceutical templates have been presented.Curcumin and its analogs,flavonoids,and marine peptides have been investigated for their possible therapeutic roles in many diseases.These compounds are promising molecular templates for radiopharmaceuticals because of their great variety of therapeutic properties and,with or without molecular modifications,they can be labeled with numerous radionuclides.Hence,successful radiolabeling of these compounds would be a powerful stimulus for radiochemists to radiolabel not only the previously mentioned compounds but also many other natural products available in nature.Therefore,this review paves the way for future exploration and development of new natural product-based radiopharmaceuticals for diagnostic and therapeutic purposes.
The methodology of radiolabeling a natural product using either direct or indirect labeling to provide a diagnostic or therapeutic agent with suitable in vivo stability and high binding affinity to the target receptor has progressed significantly in the past few years.Although considerable progress has been made,the development of natural product-based radiopharmaceuticals remains relatively challenging owing to the complex chemistry of natural products.Thus,further effort is required with respect to design,modification,and radiolabeling strategies.
The rational design of new natural product-based radiopharmaceuticals requires structural characterization to provide a comprehensive structure-activity relationship with the target molecule in the biological system and evaluate the success or failure of a radiolabeling strategy.However,from a molecular elucidation point of view,proof of structures has not been presented in some studies.Radiolabeled compounds cannot be characterized because of their small amounts.Nevertheless,the structures of the radiolabeled compounds can be predicted by their corresponding cold-labeled compounds.For this reason,cold compounds can be prepared at the macroscopic scale to allow structural elucidation using spectroscopic techniques,such as nuclear magnetic resonance spectroscopy,mass spectrometry,infrared spectroscopy,and X-ray crystallography.For example,the structural characterization of99mTc-labeled compounds can be obtained using their rhenium analogs.Additionally,the introduction of radionuclides should not alter the biological activity of the bioactive molecules;to this end,a radiolabeling strategy using the BFC approach or introducing a linker between a biomolecule and a radionuclide can be effective.
CRediT author statement
Hendris Wongso:Conceptualization,Methodology,Software,Validation,Formal analysis,Investigation,Resources,Data curation,Writing-Original draft preparation,Reviewing and Editing,Visualization.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This study was supported by the Center for Applied Nuclear Science and Technology of the National Nuclear Energy Agency of Indonesia.
 Journal of Pharmaceutical Analysis2022年3期
Journal of Pharmaceutical Analysis2022年3期
- Journal of Pharmaceutical Analysis的其它文章
- Highly sensitive electrochemical determination of rutin based on the synergistic effect of 3D porous carbon and cobalt tungstate nanosheets
- Characterization of multiple chemical components of GuiLingJi by UHPLC-MS and 1H NMR analysis
- Modifying current thin-film microextraction(TFME)solutions for analyzing prohibited substances:Evaluating new coatings using liquid chromatography
- Discussion on the dimerization reaction of penicillin antibiotics
- A strategy of screening and binding analysis of bioactive components from traditional Chinese medicine based on surface plasmon resonance biosensor
- MIL-53-based homochiral metal-organic framework as a stationary phase for open-tubular capillary electrochromatography