
Identification of soft rot resistance loci in Brassica rapa with SNP markers
2022-07-18 03:47LIUMengyangWUFangGEYunjiaLUYinZHANGXiaomengWANGYanhuaWANGYangYANJinghuiSHENShuxingZHAOJianjunMAWei
LIU Meng-yang,WU Fang,GE Yun-jia,LU Yin,ZHANG Xiao-meng,WANG Yan-hua,WANG Yang,YAN Jing-hui,SHEN Shu-xing,ZHAO Jian-jun,MA Wei
State Key Laboratory of North China Crop Improvement and Regulation,Ministry of Science and Technology/Key Laboratory of Vegetable Germplasm Innovation and Utilization of Hebei/Collaborative Innovation Center of Vegetable Industry in Hebei/College of Horticulture,Hebei Agricultural University,Baoding 071000,P.R.China
Abstract Soft rot caused by Pectobacterium carotovorum (Pc) is a devastating disease of Brassica rapa,causing substantial reductions in crop yield and quality. Identifying genes related to soft rot resistance is the key to solving this problem.To characterize soft rot resistance,we screened a soft rot-susceptible Chinese cabbage (A03),a resistant pakchoi(‘Huaguan’),and a resistant mutant (sr). An F2 population was generated by crossing susceptible Chinese cabbage A03 and resistant pakchoi ‘Huaguan’ to identify quantitative trait loci (QTLs) that confer soft rot resistance. A high-density genetic map was constructed and the three QTLs identified contain 166 genes. Based on available transcriptome data,we analyzed the expression of the 166 genes during an important defense regulatory period in Pc infection in both A03 and the resistant mutant sr. Among the 166 genes,six candidate genes were related to the soft rot defense response in B.rapa. TIFY10B (JAZ2,BraA07g038660.3C) was located in the major soft rot resistance QTL,DRQTL-3 on A07,and we speculate that this gene may play an important role in the defense mechanism against soft rot in B.rapa. This study lays the foundation for further investigations on the mechanism of soft rot resistance in B.rapa crops.
Keywords: soft rot,quantitative trait loci-sequencing,Brassica rapa
1.Introduction
Brassicarapais comprised of a number of important vegetable and oil crops,such as Chinese cabbage(B.rapaL.ssp.pekinensis),pakchoi (B.rapaL.ssp.chinensis),and turnip (B.rapaL.ssp.rapa). Soft rot caused by the pathogenPectobacteriumcarotovorum(Pc) is one of the major diseases ofB.rapacrops,and it can substantially reduce the yield of susceptible varieties(Gardanet al.2003).
Pccan invade the host plant through wounds generated by natural causes,insect bites,diseases,and mechanical injuries. It secretes plant cell-wall-degrading enzymes (PCWDEs) and absorbs nutrients from plant tissues to support its proliferation and to further infect the plant (Charkowskiet al.2012). Plants recognize pathogen-associated molecular patterns (PAMPs) through specific pattern recognition receptors (PRRs) and initiate a variety of resistance responses againstPcto trigger pattern-triggered immunity (PTI;Jones and Dangl 2006).Therefore,the initiation of plant disease resistance mechanisms requires a complex network of signals.Salicylic acid (SA),jasmonic acid (JA),and ethylene (ET)are the main signals that activate the immune response(Yanget al.2015). JA/ET-dependent signaling pathways play essential roles in conferringPcresistance,however,it is not known whether the SA-dependent pathway is also required for this process (Normansetterbladet al.2000;Kariolaet al.2003;Liet al.2004).
Little is known about the defense genes that respond to soft rot infection,and most of this information comes from studies onArabidopsisthaliana. For example,WRKY70is a key factor in the defense againstPcinA.thaliana. It functions in plant defense pathways as an inducer of SA-dependent signaling and an inhibitor of JA-dependent signaling (Liet al.2004).WRKY75also functions as a positive regulator of the JA-or SA-mediated defense responses againstPc(Choiet al.2014). Upon heterologous expression of the Chinese cabbageWRKY7(BrWRKY7) inA.thaliana,the expression of the JA-dependent signaling genePDF1.2was activated,indicating thatWRKY7is a positive regulator of the JA pathway (Koet al.2015). Furthermore,A.thalianaexpressingBrWRKY33showed resistance to soft rot(Zhenget al.2006).
Chinese cabbage is a native crop of China and one of the country’s most important vegetables. Soft rot is one of the three most economically damaging diseases of Chinese cabbage. In our previous study,plants harboring the resistant mutantsrgene,which confers resistance toPc,exhibited upregulation of the JA and ET biosynthesis genes. Furthermore,PTI was central to the plant defense response againstPcand was strongly activated at 12 hours post-infection (hpi) in thesrmutant (Liuet al.2019).However,the key resistance genes against soft rot have not been fully characterized. Plant disease resistance is controlled by multiple quantitative trait loci (QTLs),and its genetic mechanisms are complex (Dalton-Morganet al.2014). The identification of SNP loci by high-throughput sequencing and the construction of genetic linkage maps for disease resistance QTLs are needed to clarify the mechanisms of disease resistance and to permit the breeding of resistant varieties (Joneset al.2006).The whole genome resequencing method improves the resolution of linked QTLs and can accurately identify target genes,thereby significantly improving the efficiency of selection (Takagiet al.2013).
The goal of this study was to identify QTLs associated with resistance to soft rot inBrassicaspecies. Using an F2population derived from a cross between susceptible Chinese cabbage A03 and resistant pakchoi ‘Huaguan’,we mapped QTLs forBrassicasoft rot resistance through the construction of a high-density genetic map. We combined the results of QTL analysis with the expression profiling of candidate genes during an important defense regulatory period afterPcinfection and pinpointed six candidate genes for soft rot resistance inB.rapa.
2.Materials and methods
2.1.Plant materials
Thirty accessions ofBrassicarapawere collected from various germplasm banks. The A03 doubled haploid line served as the wild type (WT) and was used to create the EMS mutant library for Chinese cabbage,from which the soft rot-resistant mutantsrwas isolated (Luet al.2016;Liuet al.2019). Thirty-two accessions were sown in a greenhouse in February 2018,and the soft rot resistance was characterized when the plants grew to the 7-to-8 leaf stage (Appendix A).
The mapping population was comprised of 435 F2plants. The male parent was soft rot-susceptible Chinese cabbage A03,and the female parent was soft rotresistant pakchoi ‘Huaguan’ (Appendix B). F2plants,F1plants,and parental lines were sown in the field at the Hebei Agricultural University (Baoding,Hebei,China) in September 2018.
2.2.Characterization of soft rot resistance
The pathogenPectebaceriumcarotovorumBC1 (Liet al.2019;Chenet al.2021) was inoculated into 3-5 mL of LB medium,incubated at 28°C for 16 h at 110 r min-1,and then diluted with LB medium to 108CFU mL-1. The leaf disks were inoculated withPc,and the macerated lesions on the leaf disks were scored using a scale of 0 to 9invitroaccording to the method described by Liuet al.(2019). The disease rating criteria were as follows:0(no symptoms),1 (lignified inoculation spots),3 (discrete lesions;macerated lesions occupying less than 5% of the petiole),5 (macerated lesions occupying less than 25%of the petiole),7 (macerated lesions occupying less than 50% of the petiole),and 9 (macerated lesions occupying more than 50% of the petiole). The disease index (DI)was calculated as follows:∑nX/(N×9)×100%,whereXis the disease score,nis the number of plants with this score,andNis the total number of plants. Sample plants with the different soft rot disease severity ratings are shown in Appendix C.
2.3.DNA extraction and whole-genome sequencing
Genomic DNA was extracted from the young leaves of A03,‘Huaguan’,and the F2population (n=150) using the CTAB method (Murray and Thompson 1980;Rogers and Bendich 1985). The DNA concentration was determined using a NanoDrop 8000 instrument (Thermo Fisher Scientific,Waltham,MA,USA). After quality assessment,the DNA samples were randomly sheared with a Covaris crusher,and the DNA library was constructed using the TruSeq Library Construction Kit (Illumina Inc.,San Diego,CA,USA). The library was sequenced on the Illumina HiSeqTMPE150 platform at Novogene Bioinformatics Technology Company (Beijing,China). Raw data in the fastq format were processed and cleaned. The clean data were mapped to theB.rapareference genome(v3.0;Zhanget al.2018) using BWA Software (Li and Durbin 2009). A total of 8 127 342 SNPs from the parental genotypes (A03 and ‘Huaguan’) was detected using Genome Analysis Toolkit Software (Mckennaet al.2010).SNP variants were functionally annotated using ANNOVA Software (Wanget al.2010).
2.4.Genetic map construction and QTL mapping
Based on the selected homozygous SNPs with the same genotype as one of the parents,a sliding window was used to call genotypes at these SNP sites and to determine the recombination breakpoints. Each chromosome segment in each F2individual that was not separated by recombination breakpoints was defined as a recombinant bin,and these recombinant bins were used as genetic markers. A total of 2 494 markers was used to generate a genetic map of the F2population (n=150) covering the 10 linkage groups(Appendix D). The same banding pattern as the A03 parent was denoted with ‘a’,the same pattern as the ‘Huaguan’ parent was denoted with ‘b’,the heterozygotes were denoted with ‘h’,and unclear or missing data were denoted with ‘-’. JoinMap v4.0 Software was used to construct the genetic map,and the QTL regions for soft rot resistance were identified by interval mapping (IM) analysis using QTL lciMapping 4.1 Software. Genome-wide logarithm of odds (LOD)scores of 2 was used as threshold,and Map Charter 2.1 Software was used to draw the genetic map. Soft rot resistance QTLs were designated DRQTL-1,DRQTL-2,and DRQTL-3.
2.5.Candidate gene expression analyses
In our previous study of soft rot resistance in Chinese cabbage,we performed RNA-seq transcriptome profiling of tolerant mutantsrplants and susceptible A03 plants at 0,6,12,and 24 hpi withPcto investigate the putative molecular mechanism ofPcresistance. We found that 12 hpi was the most important defense regulatory time point duringPcinfection of Chinese cabbage (Liuet al.2019).Here,we selected genes in QTL regions for which the paralogues displayed different expression patterns in the wild type (WT) and mutantsrat 0 and 12 hpi based on the heatmap.
To analyze the expression pattern of each candidate gene,total RNA was extracted from A03,‘Huaguan’ and mutantsrat 0 and 12 hpi,and first-strand cDNA was synthesized using a FastQuant RT Super Mix (TIANGEN,Beijing,China). The gene primers were designed using Primer Premier 5.0 Software and are shown in Appendix E.
qRT-PCR analysis was performed on a Lightcycler 96 real-time PCR detection system (Roche,Basel,Switzerland) using the SYBR Green PCR Master Mix(TaKaRa,Dalian,China). The 2-ΔΔCtmethod was used to calculate the relative expression levels of the target genes (Livak and Schmittgen 2001). All reactions were performed with three biological and technical replicates.
3.Results
3.1.Characterization of soft rot resistance in 32 B.rapa accessions and an F2 population
Thirty-twoB.rapaaccessions were inoculated withPc invivo(Appendix A). Six accessions were identified as highly susceptible,nine as susceptible-to-highly susceptible,six as susceptible,four as tolerant-tosusceptible,one as tolerant,one as resistant-to-tolerant,three as resistant,and one as highly resistant-to-resistant.
The two parents,Chinese cabbage A03 and pakchoi‘Huaguan’,were inoculated withPcinvitroandinvivo,and members of the F1and F2populations were inoculated withPcinvitro(Table 1;Fig.1). The disease severity of inoculated A03 plants was scored as either 5,7,or 9 following both inoculation methods,and their DI was 70.01-80.00,indicating that they were susceptibleto-highly susceptible to soft rot. The disease severity ofinvivoinoculated ‘Huaguan’ plants was scored as either 1,3,or 5,and their DI was 23.70,indicating that they were resistant to soft rot. The disease severity ofinvitroinoculated ‘Huaguan’ plants was scored as either 1,3,5,7,or 9,and their DI was 32.22,indicating that they were resistant-to-tolerant to soft rot. The disease severity ofthe five F1plants was scored as 7 or 9invitro,indicating high susceptibility to soft rot. The disease severities of the 435 F2plants ranged from 0 to 9,and most scored between 3 and 7 (Appendix F).
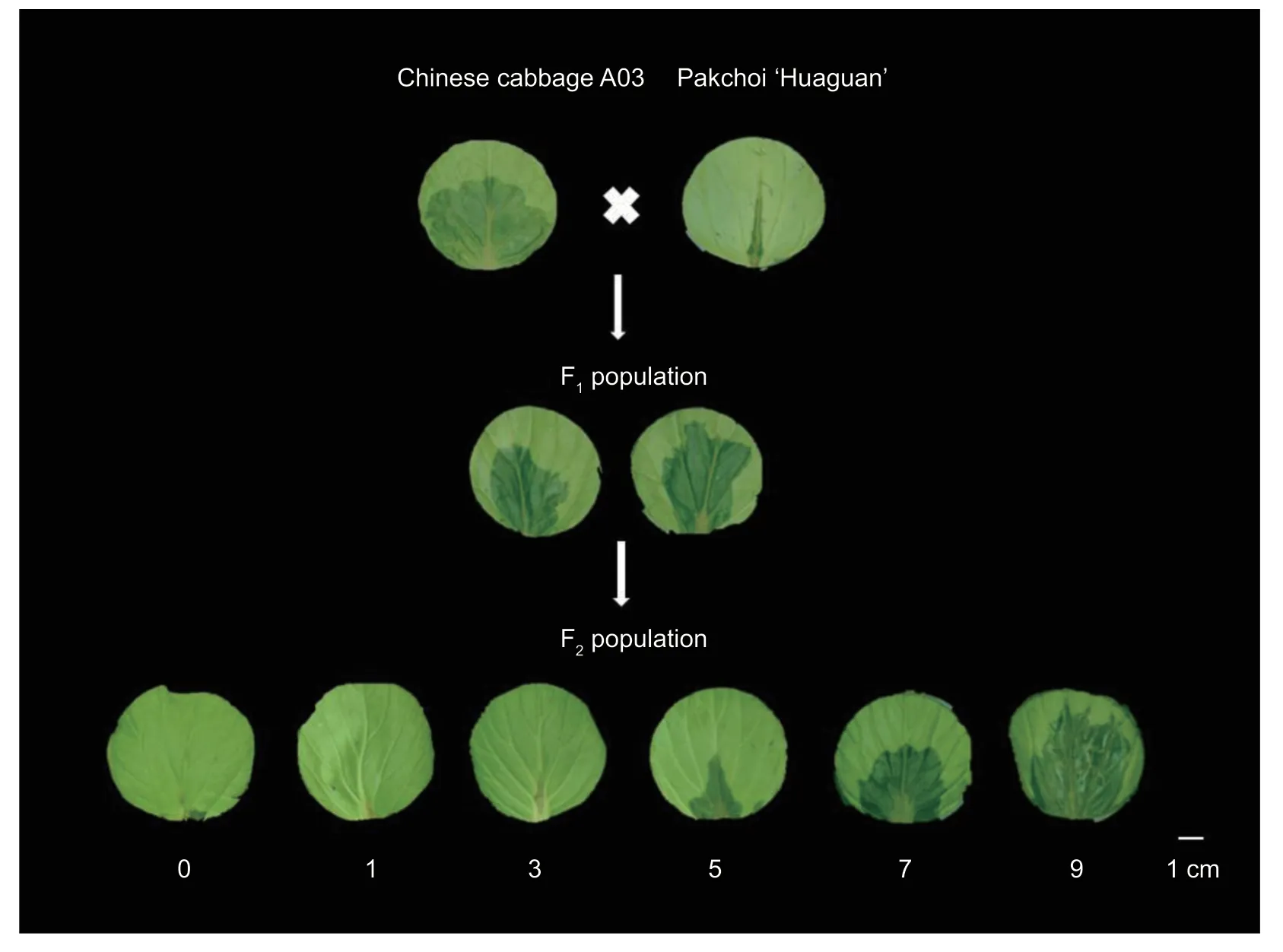
Fig. 1 The disease grading standard and disease severity for soft rot of the parents,and the F1 and F2 populations in vitro. The disease grading of the F2 population included values of 0,1,3,5,7,and 9.
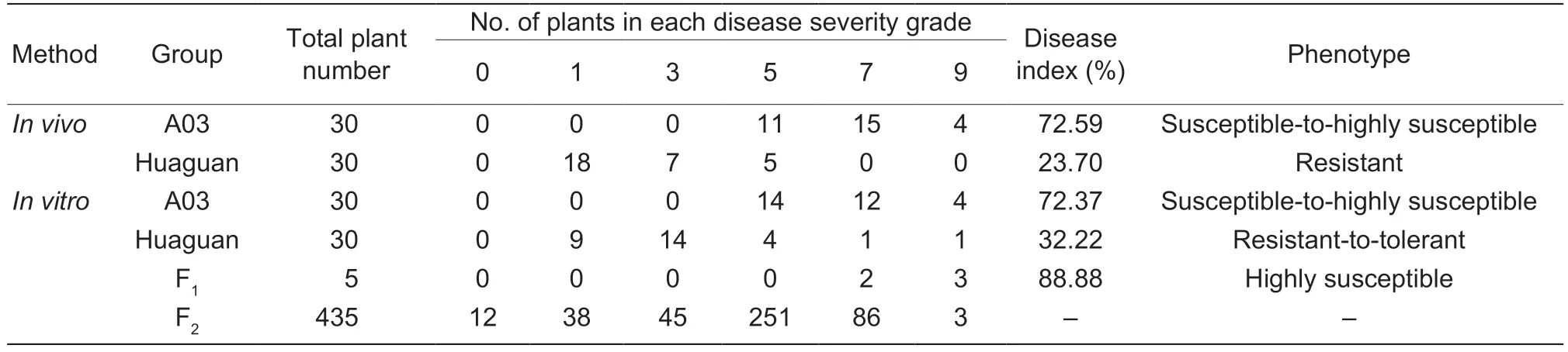
Table 1 Disease severity of soft rot in the parents,and the F1 and F2 populations
3.2.Linkage map construction
Ten linkage groups (A01-A10) were defined using 2 494 SNP markers (Fig.2;Appendix G). The length of the map was 1 773.63 cM,and the average distance between markers was 0.71 cM. The numbers of markers in the different linkage groups ranged from 171 (A08) to 322(A09),and their lengths varied from 85.57 cM (A08) to 248.06 cM (A02). Among the 10 linkage groups,the largest average spacing was 1.03 cM for A10,and the smallest average spacing was 0.50 cM for A08.
To evaluate the quality of the linkage map,we used 2 494 SNP markers to compare the genetic and physical maps (Appendix H). Only a few markers were upside down relative to their physical locations on chromosomes A01,A05,A07,and A09,indicating that the genetic map corresponded well with the physical map.
3.3.Soft rot resistance QTL identification
Interval mapping was used to identify QTLs associated with soft rot resistance in the F2population (n=150),and three resistance QTLs were detected on two different chromosomes (A02 and A07;Fig.3;Table 2). DRQTL-1 and DRQTL-2 were located on A02 (3.36-4.04 cM and 35.70-45.32 cM,respectively;LOD=2.032-2.129);their contributions ranged from 4.75 to 4.87%,and they showed positive additive effects. DRQTL-3 was located on A07 (36.55-37.22 cM,LOD=3.153) and had a contribution of 7.16%,so it was identified as the main QTL region with a positive additive effect. A total of 166 genes were identified in the three QTL regions (Appendix I).

Table 2 Soft rot resistance quantitative trait loci (QTLs) identified from the F2 population1)

Fig. 2 The genetic linkage map of the F2 population constructed by crossing Chinese cabbage A03 and pakchoi ‘Huaguan’.
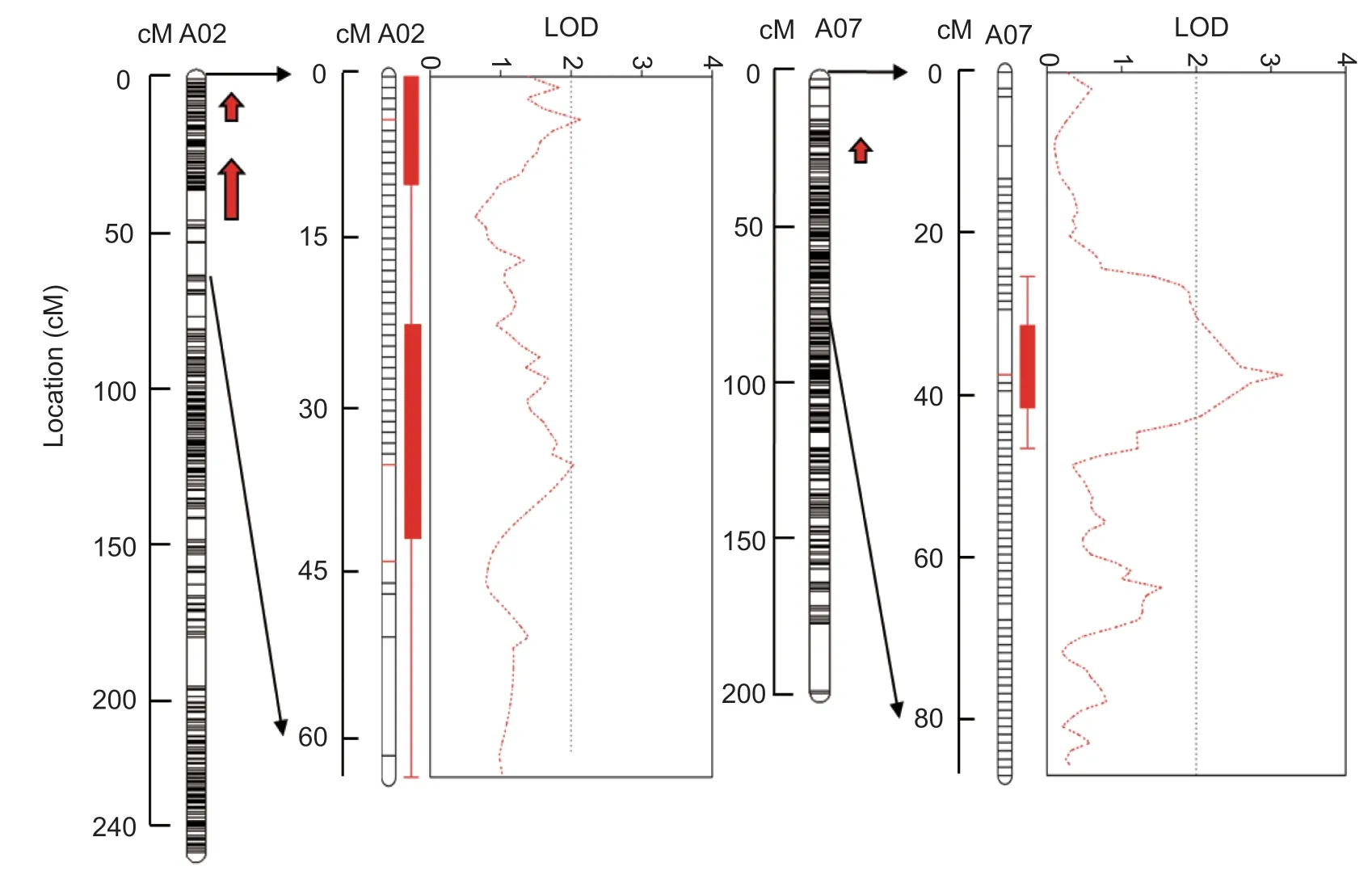
Fig. 3 The location of quantitative trait loci (QTLs) for soft rot resistance in the F2 population. For A02 and A07,left,Linkage map containing the resistance locus,with map distance indicated on the y axis;right,QTL likelihood-profile for the resistance trait,with LOD score on x axis. The length of each red arrow indicates the interval. The direction of each red arrow represents the direction of the additive effect.
3.4.Investigation of candidate gene functions
Based on available transcriptome data (Liuet al.2019),we analyzed the transcript levels of the 166 genes in susceptible Chinese cabbage A03 (WT) and the resistant mutantsrat 0,6,12,and 24 hpi. Sixty-four genes were expressed,and their expression patterns were divided into four groups by clustering analysis (Fig.4). Upon the infection of WT and mutantsrplants withPc,the expression of Groups I and II genes was downregulated in response to soft rot;specifically,their expression was unchanged at 6 hpi in either the WT or mutantsr,but decreased after 12 and 24 hpi in the mutantsrand after 24 hpi in the WT. The 14 genes in Group II and the 15 genes in Group IV were downregulated in the WT and mutantsrbut upregulated in the mutantsrat 12 hpi,suggesting that they may contribute to the immune response againstPc. Among the 29 expressed genes in Groups II and IV,seven were potentially involved in the immune response (Appendix J). Specifically,two genes (BraA07g038660.3CandBraA02g009440.3C)participated in hormone-and calcium-mediated signal transduction,four (BraA02g009410.3C,BraA02g009830.3C,BraA02g010260.3C,andBraA02g010570.3C) contributed to the defense response during pathogen infection,and one (BraA02g009880.3C)is associated with the synthesis of secondary metabolites.

Fig. 4 Heatmap of the major differentially expressed genes between susceptible A03 (wild type,WT) and the resistant mutant sr infected with Pc at 0,6,12,and 24 hours post-infection (hpi),respectively.
Our previous analysis of the putative immune mechanism of the Chinese cabbage-Pcinteraction demonstrated that 12 hpi was the most important defense regulatory time point (Liuet al.2019). Here,the expression levels of seven genes were confirmed and analyzed by RNA-seq data in the WT and mutantsrat 0 and 12 hpi,and the results showed that the trends in the two groups of data were basically the same (Fig.5).Among these seven genes,one gene from Group II(BraA02g009410.3C) was expressed at very low levels inBrassicaspecies and was therefore not considered to have a role in the defense response against soft rot.We also determined the expression levels of these six candidate genes in WT,‘Huaguan’ and mutantsrat 0 and 12 hpi (Fig.5). The two genes (BraA07g038660.3C,andBraA02g010570.3C) from Group II and four genes (BraA02g009830.3C,BraA02g009880.3C,BraA02g010260.3C,andBraA02g009440.3C) from Group IV shared a similar expression pattern,in that their expression increased at 12 hpi and was higher in the mutantsrthan in the WT (Fig.5). Even more noteworthy is that two genes (BraA07g038660.3C,andBraA02g009830.3C) were expressed significantly higher in resistant ‘Huaguan’ and mutantsrat 12 hpi. These genes may have exerted positive roles in the defense response againstPc.
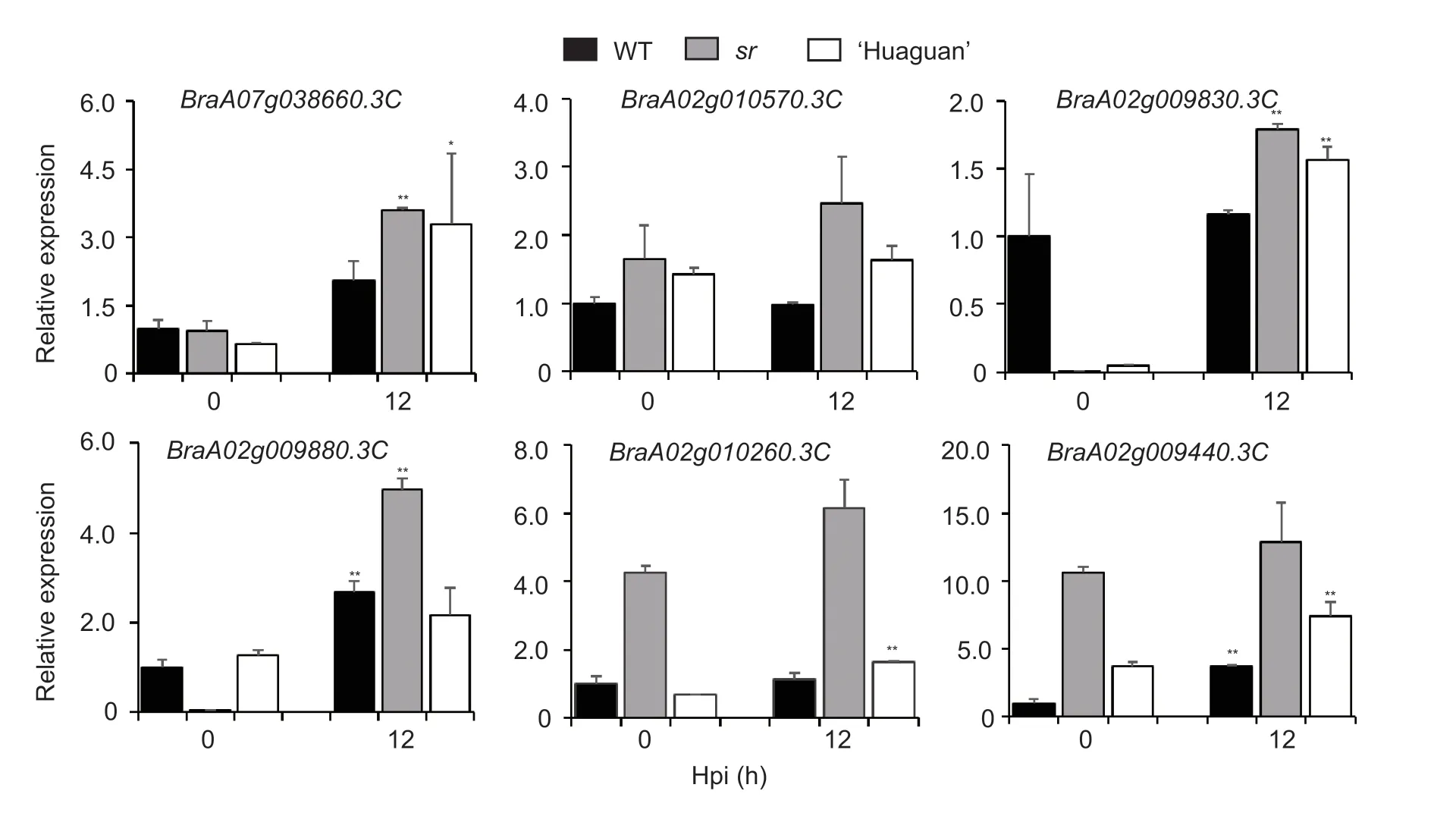
Fig. 5 The relative expression levels of six genes in susceptible A03 (wild type,WT),resistant ‘Huaguan’ and mutant sr and at 0 and 12 hours post-infection (hpi). **,P<0.01;*,P<0.05 (n=3,Student’s t-test).
Taken together,these six annotated candidate genes were from two QTL regions and were potentially related to soft rot resistance (Table 3). Five genes (BraA02g010570.3C,BraA02g009830.3C,BraA02g009880.3C,BraA02g010260.3C,andBraA02g009440.3C) were located in DRQTL-2 on A02(35.70-45.32 cM,LOD=2.032,contribution 4.75%);and one (BraA07g038660.3C) in DRQTL-3 on A07,which had the strongest positive effect onPcresistance.

Table 3 Annotation of the six candidate genes related to soft rot resistance1)
4.Discussion
Soft rot is one of the major diseases ofB.rapa. It can cause plant rot and spread across a large area from spring to autumn,substantially reducing the yield ofB.rapacrops. Exploring soft rot resistance genes,uncovering the molecular mechanisms of soft rot resistance,and cultivating highly-resistant varieties is the most effective strategy for combatting soft rot. In this study,we identified six candidate genes that may be potentially related to soft rot resistance,and our findings improve our understanding of the mechanism of soft rot resistance.
4.1.Construction of a high-density linkage map and identification of candidate resistance genes
The construction of a linkage map with molecular markers is a key step in the linkage analysis of important biological or agronomic traits (Horiet al.2003). A high-density linkage map can be used to accurately identify soft rot resistance genes (Huet al.2018). The selection of parental lines is important when constructing linkage maps,as it directly affects the difficulty and scope of application (Wanet al.2007). Therefore,high polymorphism and purity should be taken into account when selecting the parents (Zhanget al.2005). We constructed a high-density linkage map from an F2population derived from Chinese cabbage A03 and pakchoi ‘Huaguan’ as parents. These two accessions have markedly different degrees of soft rot resistance,laying the foundation for the construction of a high-quality genetic linkage map. In previous disease resistance studies ofB.rapa,InDel,SSR,RAPD,AFLP,SRAP,and SNP markers have typically been used to construct the genetic maps (Liet al.2007). With the advent of wholegenome sequencing (Huanget al.2012),a large number of SNP markers can be used to construct high-density genetic maps,thereby providing new guidance for the identification of soft rot resistance genes inB.rapa. In this study,10 linkage groups were defined based on 2 494 SNP markers,which were useful for soft rot QTL analyses.
According to the published data,soft rot resistance is controlled by multiple QTLs (Yamagishiet al.1990;Zimnoch-Guzowskaet al.2000). Several genes have been demonstrated to enhancePcresistance,including genes encoding pineapple bromelain (BAA1;Junget al.2008),rice leucine-rich repeat protein (OsLRP;Parket al.2012),polygalacturonase-inhibiting protein 2 (PGIP2;Hwanget al.2010),WRKY7(Koet al.2015),WRKY12(Kimet al.2014),WRKY33(Zhenget al.2006),WRKY70(Liet al.2004),andWRKY75(Choiet al.2014). In our previous study,we compared the transcript profiles of resistant mutantsrplants and susceptible WT plants at 0,6,12,and 24 hpi in response toPc(Liuet al.2019).Putative immune mechanisms related to PTI were strongly activated insrplants at 12 h afterPcinoculation.Among the 166 genes within the three QTL regions,we identified six candidate genes related to soft rot resistance by analyzing gene expression in susceptible A03 and the resistant mutantsr. These six genes from three QTLs may contribute to the defense response against soft rot inB.rapa.
4.2.Candidate soft rot resistance genes within the QTLs
Putative molecular resistance mechanisms againstPcinclude pathogen recognition,signal transduction,and the synthesis of secondary metabolites that function in the immune response (Liuet al.2019). In general,plants activate the immune response againstPcviapathogen recognition and signal transduction. When PRRs on the surface of cell membranes recognize M/PAMPs or DAMPs,the downstream defense response strongly activates the Ca2+-mediated resistance response,thereby activating calcium-dependent protein kinases (CPKs) and calcium-binding proteins (CMLs;Liuet al.2019). The candidate geneCAM7(BraA02g009440.3C) is a calcium sensor that interacts with the transporterPEN3and plays an important role in non-host resistance inA.thaliana(Campeet al.2016),emphasizing the importance of Ca2+sensors in the plant immune response.
The WRKY proteins are one of the largest families of plant transcription factors,and they have a conserved WRKY domain and regulate plant responses to pathogens(Eulgem and Somssich 2007). Five of them,WRKY7,WRKY12,WRKY33,WRKY70,andWRKY75(Liet al.2004;Zhenget al.2006;Choiet al.2014;Kimet al.2014;Koet al.2015;Liuet al.2019),have been reported to be downstream genes forPcresistance in plants.WRKY38,which encodes a type III WRKY transcription factor,was identified as a direct target ofNPR1(Wanget al.2006);however,it is a negative regulator of disease resistance inA.thalianaduring pathogen infection (Kim and Chen 2006). The expression ofWRKY38(BraA02g009830.3C)was upregulated at 12 hpi in the resistant mutantsr. In the plant defense response,one gene may play several roles within the signaling network. WhetherWRKY38plays a positive role in the response to soft rot in plants is yet to be determined.
Plant hormones serve as signals that mediate the defense responses to pathogens and phytophagous insects. SA,JA,and ET signaling pathways activate and facilitate plant immune responses (Yanget al.2015).JAs,including jasmonic acid and methyl jasmonate(MeJA),are lipid-derived hormones that regulate plant development and responses to biotic and abiotic stresses(Howe 2018). Furthermore,JA/ET-dependent signaling pathways promote resistance to necrotrophic pathogens(Mengiste 2012). JA accumulation is involved in the immune response of Chinese cabbage toPc,which stimulates host plants to increase the production of endogenous JA,thereby significantly increasing the expression of related genes in the resistant mutantsr(Liuet al.2019). Therefore,JA-mediated signaling may be the most significant againstPcinfection in Chinese cabbage(Chenet al.2021). The candidate geneTIFY10B(BraA07g038660.3C),also known asJAZ2,belongs to the JAZ protein subfamily. JAZs are key transcriptional inhibitors of JA signaling (Chiniet al.2007;Thineset al.2007). JAZs interact withMYC2and suppress its activity,thereby negatively regulating the expression of the JA-mediated defense response genesPDF1.2,CHIB/PR3,andHEL/PR4,and ultimately regulating the invasion ofBotrytiscinerea,Fusariumoxysporium,andPseudomonadaceae(Lorenzoet al.2004;Gimenez-Ibanezet al.2017). In addition,many other factors,such as WRKY (WRKY33andWRKY57) and NPR (NPR3,NPR4) in the SA signaling pathway (Howeet al.2018),can directly interact with JAZs to affect the disease resistance of host plants. In cotton,the overexpression ofGhJAZ2decreases the expression of JA-response genes and enhances the susceptibility toVertiiumdahliaeand insect herbivory (Heet al.2017). Similarly,in our study,the expression levels ofTIFY10B(JAZ2) were significantly higher at 12 hpi in the resistant ‘Huaguan’ and mutantsrthan in the WT (Fig.5). Moreover,it was located in DRQTL-3 on A07,which was the strongest resistance QTL identified in the present work. We speculate thatTIFY10Bmay play an important role in the defense response against soft rot inB.rapa. The genetic variation sites ofTIFY10Bare shown in Appendix K providing more information for the breeding of resistant cultivars.
5.Conclusion
We constructed an F2population by crossing susceptible Chinese cabbage A03 and resistant pakchoi ‘Huaguan’and used it to identify candidate soft rot resistance genes.By combining the results of QTL mapping and transcript profiling,we identified six candidate genes that may be involved in the defense mechanism against soft rot.These results provide new insights into the molecular mechanisms of soft rot resistance inB.rapaand guidance for the breeding of resistant cultivars.
Acknowledgements
The authors are grateful to Prof.Xie Hua (Beijing Agro-Biotechnology Research Center,Beijing Academy of Agriculture and Forestry Sciences,China) for the gift of thePcpathogen. This work was supported by the National Natural Science Foundation of China (31672151 and 31902005),the China Postdoctoral Science Foundation(2019M651059),the Natural Science Foundation of Hebei,China (C2020204111),the International Cooperation Project in the Science and Technology Support Program of Hebei,China (2019YX023A),the International Cooperation Base Project in the Technology of Hebei,China (20592901D),100 Foreign Experts Plan of Hebei Province and the Starting Grant from Hebei Agricultural University,China (YJ201955),the Science and Technology Research Project of Universities in Hebei Province,China (QN2021074).
Declaration of competing interest
The authors declare that they have no conflict of interest.
Appendicesassociated with this paper are available on http://www.ChinaAgriSci.com/V2/En/appendix.htm
 Journal of Integrative Agriculture2022年8期
Journal of Integrative Agriculture2022年8期
- Journal of Integrative Agriculture的其它文章
- Historical trends in maize morphology from the 1950s to the 2010s in China
- The rhizosphere microbial complex in plant health:A review of interaction dynamics
- Allele mining of wheat ABA receptor at TaPYL4 suggests neofunctionalization among the wheat homoeologs
- Characterization of chromosome segment substitution lines reveals candidate genes associated with the nodule number in soybean
- Transcriptional profling between yellow-and black-seeded Brassica napus reveals molecular modulations on flavonoid and fatty acid content
- An economic and viable approach to improve wheat quality in Qinghai–Tibetan Plateau,China