
细胞穿膜肽识别问题的多特征融合卷积网络预测算法
2022-07-15 08:09:50周丰丰牛甲昱
电子科技大学学报 2022年4期
周丰丰,牛甲昱
(1. 吉林大学计算机科学与技术学院 长春 130012;2. 吉林大学符号计算与知识工程教育部重点实验室 长春 130012)
自从30 多年前第一个细胞穿膜肽(cell penetrating peptides, CPPs)被发现以来,CPPs 已经在医学领域得到了广泛应用[1]。CPPs 具有能够携带各种物质(包括小分子物质和蛋白质等)直接进入细胞而不会显著损伤细胞膜的特性[2]。这种特性使它们可以成为将物质转运到细胞中的药物递送剂,目前已被制药公司应用于多种治疗分子的局部和全身给药的临床实验中[3]。因为CPPs 的实用性,识别CPPs 也成为了一个有价值的研究领域。
传统的实验方法费时费力,当前生物序列预测领域采用更加省时高效的计算方法对序列进行预测[4-5]。目前,已有不少识别CPPs 的算法模型,其中大多基于机器学习算法。如文献[6]鉴定了111 种已知的CPPs 和34 种已知的non-CPPs,并使用基于不同生化特性的特征表示算法和支持向量机(support vector machine, SVM)对CPP 进 行 分类。文献[7]提出了一种同样基于SVM 的预测方法CellPPD,它的改进之处在于使用了更多的特征表示算法和更大的数据集。文献[8]提出的C2Pred使用基于二肽的特征提取方法,降维后用SVM 分类器对CPP 进行了分类,也取得了很好的结果。文献[9]构建了一个基于随机森林分类器的两层预测模型,该模型不仅可以对CPPs 和non-CPPs 进行分类,还可以预测CPPs 的吸收效率高低。文献[10]开发了CPPred FL,该模型使用了19 种不同的特征表示算法和多种不同的机器学习分类器。
最近也有一些研究者使用深度学习方法来进行CPPs 的识别,如文献[11]基于Transformer 模型提出了CPPFormer,该算法根据CPPs 序列较短的特征重构了Transformer 网络模型,并结合了多种基于生化特性的特征表示算法进行分类。文献[12]提出了DeepCPPred,是一种基于多级深度神经网络的两层预测模型。
目前基于不同的蛋白质序列特征编码算法已经有了多种识别方法,识别精度也在逐步提高。本文提出了一种基于卷积神经网络的特征表示算法ConvCPP,获得了更好的特征表示能力。ConvCPP的创新之处在于首次使用了基于卷积神经网络的算法进行细胞穿膜肽的特征表示工作,且在卷积神经网络加入了注意力模块以更好地提取序列特征。算法的主要流程为将蛋白质序列中的氨基酸编码为不同的向量,然后将编码后的序列输入到卷积神经网络中,提取网络的最后一层作为特征表示。最后,将卷积网络提取到的特征和一些传统特征表示方法得到的特征相结合,并集成了多种分类器得到最终的结果。实验结果表明,ConvCPP 在SN、SP、ACC、MCC 这4 项指标上分别达到了0.950、0.935、0.943 和0.885,在预测精确度ACC 上相对当前主流分类算法有2.2%的提升,具有更好的分类性能。本文将模型做成了CPPs 预测软件包,可在网址链接:https://pan.baidu.com/s/1Lx60bAQe_MfFa0QDKJ_rcw?pwd=hilb 下载使用。
1 细胞穿膜肽识别算法流程
1.1 模型框架
ConvCPP 的流程如图1 所示。其中,特征表示分为两部分,一部分是经过改进的卷积神经网络,另一部分是基于蛋白质理化性质的传统特征表示算法,每部分得到的特征向量都使用了T 检验进行特征选择。在分类算法部分,本文使用了朴素贝叶斯(naive Bayes, NBayes)、随机森林(random forest, RF)、 SVM、 K-最 近 邻 算 法 (K-nearest neighbor, KNN)和极限梯度推进算法(extreme gradient boosting, XGBoost)进行分类,并用投票方式集成其结果作为最终分类结果。

图1 ConvCPP 模型框架
1.2 基于卷积神经网络的特征表示算法
卷积神经网络最初被发明应用于计算机视觉领域,后来逐渐在自然语言处理领域得到了广泛应用[13],鉴于蛋白质序列和文本序列在形式上的相似性,考虑用卷积神经网络来进行特征提取。本文模型结构参考了TextCNN 的标准结构,并在其基础上进行了改进。
首先,需要对蛋白质序列中每个氨基酸进行编码,使用了随机编码的方式将20 种氨基酸编码为一维向量,设其长度为d,则长度为s的蛋白质序列可被表示为s×d维的向量矩阵。
在将向量矩阵输入卷积层之前,加入了注意力机制来更好地获取序列的位置信息。当前注意力机制在序列数据处理领域已经得到了广泛应用[14-16],本文使用的是多头注意力机制,表示如下:

式中,W0是权重矩阵;hi表 示第i个注意力头的计算结果,每个注意力头都是一个拥有不同权重矩阵的自注意力计算流程。其计算流程如下:首先,每行输入向量xi, 分别和3 个权重矩阵Wq,Wk,Wv相乘,得到向量qi,ki,vi。 之后计算注意力权重ai,公式如下:

之后对a1进行Softmax 归一化,最后计算得到最终的输入向量zi:

注意力层运算完毕之后,将结果输入卷积层。在卷积层,模型使用了多个卷积核,卷积核的个数可以作为超参数进行调整,卷积核的宽度固定为输入向量矩阵A的 宽度d,不同的卷积核可以设置为不同的高度。设卷积核w的 大小为h×d,则卷积操作的输出oi可以表示为:
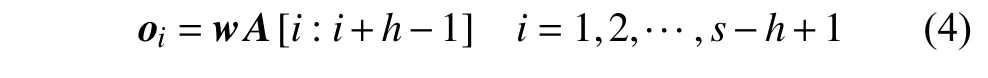
式中,A[i:i+h−1]表 示输入向量矩阵A的 第i行到第i+h−1行 。之后叠加偏置b, 经过激活函数f得到特征图c,其中:

在池化层,经典结构使用的是1-max pooling方法,即每个卷积核只取特征图中的最大值,最后将每个卷积核所得的值拼接,得到最终的特征向量。但是这种方法只取最大值,容易丢失信息,因此,本文将池化层改为动态k-max pooling 方法。这种方法根据序列的长度不同,动态保留前k个最大值,尽可能的保留了序列的特征信息。
整个卷积神经网络模型结构如图2 所示,模型共包含一个注意力层、一个卷积层和一个池化层,池化层的结果作为序列的特征输出。
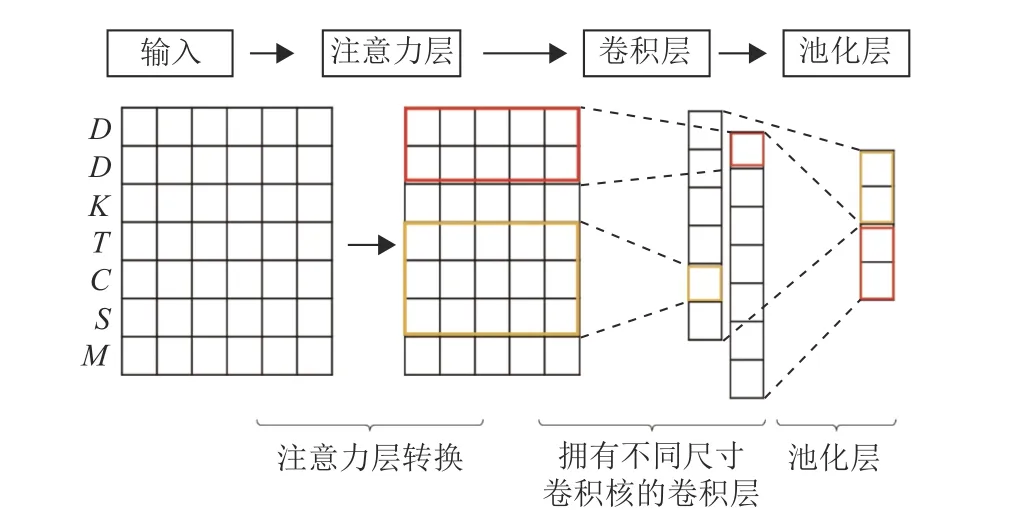
图2 卷积神经网络模型结构
1.3 基于蛋白质理化性质的特征表示算法
除了利用卷积神经网络得到的特征,本文还结合了3 种利用蛋白质理化性质的特征表示算法。
1)氨基酸组成(amino acid composition, AAC)是最简单且常用的蛋白质序列特征表示算法之一[10]。此算法主要利用了每种类型的氨基酸在蛋白质序列中出现的频率,如下所示:

式中,n1表 示第i种氨基酸在蛋白质序列中出现的频率,即出现次数除以序列的长度。对于给定的蛋白质序列,通过分别计算其中20 种不同氨基酸的频率,可以得到一个20 维的特征向量。
2)重叠属性表示算法,文献[17]发现每种氨基酸通常具有不只一种物理化学性质,因此,该文根据10 种理化性质将标准氨基酸分为10 组,每组可能有重叠。用10 维的0/1 向量对每个氨基酸进行编码,其中向量的每个位置代表一个氨基酸组。如果氨基酸属于相应的氨基酸组,则向量的位置设置为1,否则设置为0。最后,对长度为L的蛋白质序列进行编码,得到1 0×L大小的特征向量。
3) 20 位表示算法,文献[7]将20 种氨基酸分别用one-hot 编码方式编码,则每个氨基酸可以用20 维的一维向量来表示,则一条长度为L的蛋白质序列可以表示为一个2 0×L的二维向量。
2 实验结果与分析
2.1 实验环境和模型参数
在实验中,本文使用的Python 版本为3.6.13,Pytorch 框架的版本为1.3.1,numpy 库的版本为1.19.2,scipy 库的版本为1.5.4,pandas 库版本为1.1.5。实验使用的显卡型号为NVIDIA Tesla P100,16 GB,驱动版本为450.80.02,CUDA 版本为11.0。
本文进行特征提取的卷积网络超参数的取值设置参见表1。

表1 模型超参数设置
2.2 数据来源及评价指标
为了便于与现有方法进行比较,本文使用了最常用的CPPs 数据集CPP924[18],包含924 个蛋白质序列,其中包括462 个CPPs 和462 个non-CPPs。CPP924 数据集中的数据都来自目前最大的CPPs数据集CPPsite v2.0,且其中任意两个序列的序列相似性均小于80%。这一点非常重要,因为过高的序列相似性可能会使模型的预测性能表现受到影响。
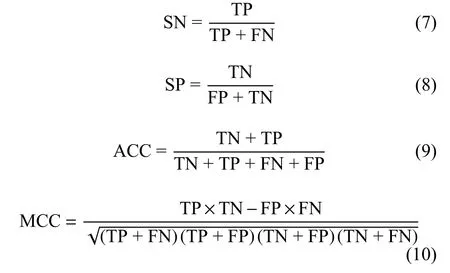
本文用到的评价指标包括敏感性值(sensitivity,SN)、特异性值(specificity, SP)、准确率(accuracy,ACC)和马修斯相关系数(Mathew’s correlation coefficient, MCC)。其计算公式分别如下:
2.3 卷积神经网络模型的消融实验
为了验证本文对卷积神经网络模型改进的有效性,本文做了消融实验来验证各模块的必要性。图3 展示了消融实验的结果,将改进分为注意力机制模块和池化层改进模块,其中模型1 表示引入注意力机制模块和池化层改进模块的模型,模型2 表示只引入注意力机制模块的模型,模型3 表示只引入池化层改进模块的模型,模型4 表示未经修改的原始模型。实验数据采用10 倍交叉验证获得。4 组实验考察了注意力模块和池化模块的不同组合方式。
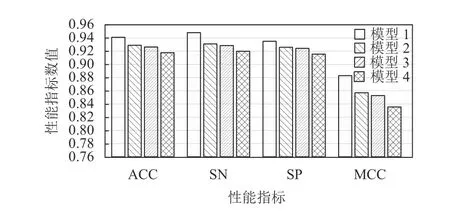
图3 模型消融实验结果
图3 的数据表明,注意力模块和池化模块对卷积神经网络原始模型具有较好的性能提升,且二者联合使用会对原始模型的改进程度更大。注意力模块将原始模型准确率(ACC)提升了0.011,而池化模块将原始模型准确率(ACC)提升了0.009。本文提出的ConvCPP算法整合了注意力模块和池化模块的优势功能,将原始模型的准确率提升了0.023。原始模型的马修斯相关系数(MCC)也提升了0.047。
2.4 不同特征表示方法的性能对比
为了验证两种特征表示方法都是有效的,本文分别将只用卷积网络得到的特征和只用利用理化性质算法得到的特征进行分类,实验结果如图4 所示。其中特征集1 包括两种方法得到的特征,特征集2 表示用卷积网络得到的卷积特征,特征集3 表示利用理化性质算法得到的理化特征。图4 显示特征集1 的结果在4 个指标上都是最好的,表明两种方法得到的特征都是有效的,而特征集2 取得的准确率(ACC)优于特征集3 至少0.016。这表明本文提出的卷积特征提取算法相对于利用理化性质的特征表示算法,能更好地利用序列中各氨基酸之间的内在关联信息来发现CPP 特异的序列特性。实验数据采用10 倍交叉验证获得。本实验考察了从序列数据中提取出来的理化特征和卷积特征对CPP 预测问题的性能贡献。
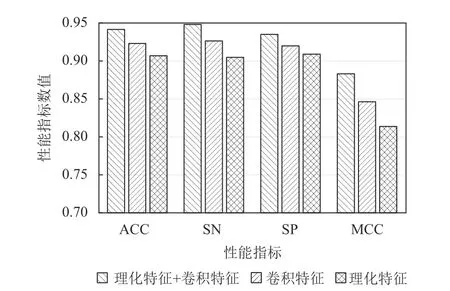
图4 不同特征集合的实验结果
2.5 不同分类器的实验结果对比
本文使用5 种分类器,通过投票方式集成为最终分类结果。这5 种分类器包括NBayes[19]、RF[20-21]、SVM[22]、KNN[23-24]、和XGBoost[25]。每种分类器各自的性能结果以及集成后模型的分类结果如表2 所示。可以看出,SVM 和XGBoost 是效果相对较好的分类器,均取得了0.930 以上的准确率(ACC),后续如需优化分类器数量可保留这两个分类器。其中XGBoost 取得了最好的准确率0.938,而集成后的分类精确度指标是最优的(ACC=0.942),比性能最好的独立分类器XGBoost 的准确率还要高0.006。实验结果说明,本文采用的投票集成法可以有效利用优势互补的各个分类器预测结果。
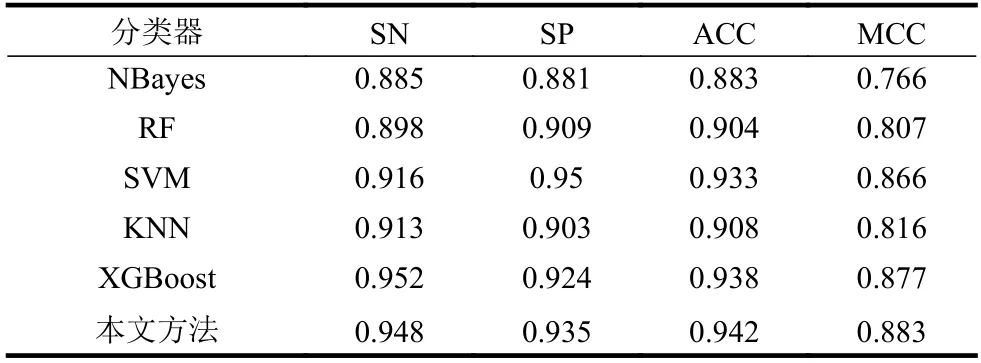
表2 不同分类器的结果对比
2.6 特征选择结果对比
为了消除两种不同特征提取方式的信息冗余,本文采用了最大相关−最小冗余(mRMR)和T 检验两种不同的特征选择算法,分别对拼接后的特征向量进行特征选择,实验结果如图5 所示,使用mRMR 算法进行特征选择,特征数为125 时得到了最优分类效果(ACC=0.943),达到了消除冗余和优化分类结果的目的。实验数据采用10 倍交叉验证获得。本实验测试了两种不同特征选择算法对分类准确率的影响。
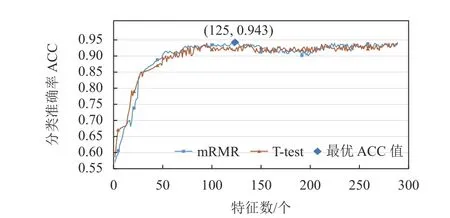
图5 两种不同特征选择算法的实验结果
2.7 与其他模型的结果对比
为了证明本文提出的ConvCPP 算法的有效性,本文和已发表的其他CPPs 预测模型的结果进行了对比。这些模型都是基于CPP924 数据集进行的预测,包括CPPred-RF[9]、文献[26]提出的算法模 型、SkipCPP-Pred[18]、CellPPD 和CPPred-FL。其中,CellPPD 根据不同的特征提取算法被分为3 个不同的模型,分别称为CellPPD-1、CellPPD-2 和CellPPD-3。实验的验证方法均为十折交叉验证,验证指标包括敏感性值(SN)、特异性值(SP)、准确率(ACC)和马修斯相关系数(MCC)。表3 给出了4 个分类性能指标。可以看出,本文提出的ConvCPP 模型在4 个指标上,都优于其他模型。准确率ACC 至少被改进了0.022,综合性能指标MCC 也至少被改进了0.043。足以证明本文算法模型的有效性与精确性。
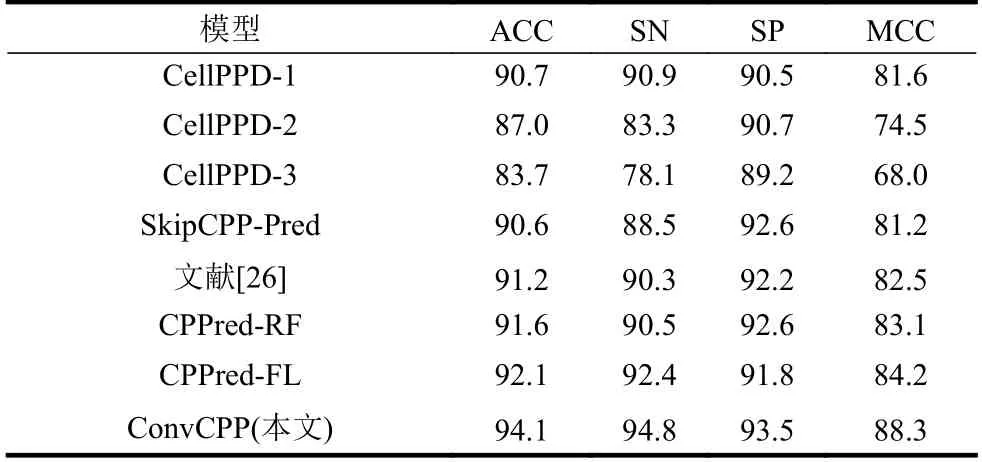
表3 与其他模型的结果对比 /%
3 结 束 语
本文提出了一种基于卷积神经网络的细胞穿膜肽识别算法ConvCPP,结合了引入注意力机制的卷积神经网络和利用蛋白质理化性质的两种特征表示方法获取的蛋白质序列特征,并结合了多种分类器进行了最终分类。经实验验证,ConvCPP 在各项指标上都达到了最佳结果,预测准确率ACC相对当前其他分类算法提升了2.2%。相关实验结果表明,深度卷积网络可以有效提取蛋白质序列的隐含模式信息,具有优秀的对细胞穿膜肽的检测能力。
猜你喜欢
小雪花·成长指南(2022年1期)2022-04-09 18:39:41
北京航空航天大学学报(2021年9期)2021-11-02 08:24:26
电子制作(2019年11期)2019-07-04 00:34:38
北京航空航天大学学报(2018年1期)2018-04-20 06:38:17
电子测试(2018年1期)2018-04-18 11:52:35
传媒评论(2017年3期)2017-06-13 09:18:10
光学精密工程(2016年4期)2016-11-07 09:05:00
光学精密工程(2016年3期)2016-11-07 09:03:33
第二课堂(课外活动版)(2016年2期)2016-10-21 16:58:54
电测与仪表(2014年15期)2014-04-04 12:05:20
