
玉米株高QTL的定位及主效QTL的确认
2022-07-05 01:10:08媛3张红伟
核农学报 2022年8期
高 歌 杨 媛3, 郑 军 张红伟,*
(1 吉林农业大学农学院, 吉林 长春 130118;2 中国农业科学院作物科学研究所,北京 100081;3 中国农业大学农学院, 北京 100193)
株高是作物重要的株型性状。玉米株高与玉米的产量和抗倒伏等性状密切相关,培育合适株高的玉米品种对于玉米的高产稳产具有重要意义[1-2]。朱永兴等[3]利用122份玉米杂交组合作为试验材料,通过分析杂交种的重要农艺性状发现,株高与产量的相关性最大。株高过高会增加作物倒伏的风险,过低则影响生物量,降低光合产物的积累,最终导致产量降低,因此,适宜的株高有助于玉米高产[4-6]。
玉米株高是一个由多基因控制的数量性状,遗传机制复杂且易受环境影响[7]。研究影响玉米株高变异的遗传基础,是精细定位株高数量性状位点(quantitative trait locus, QTL)和挖掘候选基因的铺垫工作。基于双亲群体的QTL定位是检测作物遗传变异的有效途径[8-10]。随着分子技术水平的提高及数量性状定位方法的改进,研究者们已鉴定到大量控制株高的QTL和基因。例如,Zhou等[11]利用Ye478×Qi319(Y/H)群体鉴定到14个株高相关QTL,并筛选到1个可能调控株高的候选基因GRMZM2G325907。Du等[12]利用2个加倍单倍体(doubled haploid, DH)群体,基于高密度遗传连锁图谱,鉴定到22个株高相关的QTL,其中qHT_YZ5a位于第5号染色体147.4~217.3 Mb区间,可解释11.6%~15.3%的株高和穗位高变异。同样,Li等[13]基于连锁图谱,在多个环境下共鉴定到21个控制株高和穗位高的QTL,并利用近等基因系(near isogenic lines, NIL)群体验证了位于第1号染色体上的QTL。虽然前人对株高的遗传机制研究已经取得了一定进展,但是利用不同亲本组配的分离群体检测到的QTL位置不同,因此仍需构建不同群体,对株高进行进一步解析。
全基因组关联分析(genome-wide association study, GWAS)是基于连锁不平衡的原理,在全基因组范围内检测表型性状和单核苷酸多态性(single nucleotide polymorphism, SNP)标记之间关系的分析方法[14-15],该方法能以较高的分辨率和灵敏度识别出目标性状关联的候选基因[16-17]。近年来,GWAS已广泛应用于不同作物复杂性状的遗传机制研究。Liu等[18]对236个玉米不同自交系进行全基因组关联分析,发现73个与玉米粗缩病抗性相关的SNPs,并在连锁不平衡(linkage disequilibrium, LD)区域内鉴定出41个候选基因。Zhang等[19]通过GWAS在大豆中鉴定到11个与开花天数、成熟度和株高相关的候选基因。以上研究表明GWAS是实现对作物性状进行QTL定位的有效方法。
利用生产中主流栽培种亲本构建的群体进行株高QTL定位,能够反映出主流栽培种中株高的遗传变异规律,具有重要的应用价值。因此,本研究拟利用优良杂交种光玉335的父本PH4CV,与杂交种郑单958的母本郑58共同构建的481个BC1F3∶4家系为研究材料,结合两年两点4个环境下的株高表型和基因型,进行基于混合线性模型(mixed linear model, MLM)的GWAS分析,旨在筛选出玉米株高相关QTL位点,并对主效QTL进行验证,为株高QTL的进一步精细定位奠定基础。
1 材料与方法
1.1 试验材料
本研究所用试验材料是以先玉335的父本PH4CV自交系为轮回亲本,与郑单958的母本郑58杂交获得的F1代,后与轮回亲本PH4CV进行回交,构建BC1F1代回交群体,再经过不断自交,最终得到481个 BC1F3∶4家系[20]。
1.2 材料种植与表型鉴定
包含481份家系的BC1F3∶4群体于2016和2017年分别在北京及新疆进行种植,2年2点共计4个环境,分别以16BJ、16XJ、17BJ、17XJ命名,其中BJ表示北京、XJ表示新疆,16和17分别表示2016和2017年。在以上4个环境中,均设计2次重复,并以完全随机的方式,每个家系播种单行区,单粒播种,行长5 m,每行21粒,株行距分别为25、60 cm。于散粉结束后人工测量玉米株高。
1.3 株高表型分析
利用混合线性模型计算最佳线性无偏预测值(best linear unbiased estimation, BLUE)[20],用 Excel 2019对测得的株高表型数据进行整理和分析,得到群体株高的最大值、最小值、平均值、标准差、变异系数等基本统计量。
1.4 群体基因型鉴定
利用改良的十六烷基三甲基溴化铵(cetyltrimethylammonium bromide, CTAB)方法从481份BC1F3∶4群体材料中分离出高质量的基因组DNA,通过NanoDrop2000超微量分光光度计(ThermoFisher,美国)对其浓度及纯度进行检测,再抽取部分样品用1%琼脂糖凝胶电泳进行鉴定,鉴定到的DNA浓度在20 ng·μL-1以上,纯度指标OD280/OD260值为1.70~1.90,且电泳检测到单一条带,亮度较高无杂带,即样品符合基因型鉴定标准,送至北京博奥晶典生物技术有限公司利用基因芯片进行SNP基因分型[21]。由于基因型数据的准确性对后续的关联分析有较大影响,通过一系列筛选过滤条件对SNP原始的分型结果进行严格质控:去除在群体中缺失率大于5%的标记,保留检出率大于97%的位点,去除在两亲本间无多态性差异的位点,去除最小等位基因频率小于0.05的标记位点。最终获得11 781个在基因组上均匀分布的高质量SNP标记及对应群体材料的基因型数据[20]。
1.5 混合线性模型分析
在R语言中利用lme4包计算群体的BLUE值,结合基因芯片鉴定得到的基因型数据,利用R语言中的sommer_3.5包进行基于混合线性模型[22]的GWAS分析,以错误发现率(false discovery rate, FDR)为0.05时的P值确定阈值来鉴定显著相关的SNP标记。最后用R语言qqman软件包绘制曼哈顿图(Manhattan plot)和Q-Q图(Q-Q plot)。
1.6 显著位点效应分析
依据线性回归模型,对关联分析筛选到的显著SNP标记进行表型变异率(phenotype variance explained, PVE)计算,保留PVE在1%以上的标记。先计算每个显著标记的平方和,依次相加得到标记总体的平方和,单个标记的平方和除以标记的总平方和即获得每个显著标记的PVE[20]。假定1表示轮回亲本PH4CV的基因型,0表示杂合的基因型,-1表示供体亲本郑58的基因型,标记的加性效应值为基因型为1的家系株高表型平均值与基因型为-1的家系表型平均值间差值的一半。
1.7 基因型验证
以PH4CV为轮回亲本构建得到BC1F1群体,经不断自交获得具有大量分离表型的BC1F5:6家系群体。在效应最大的标记上下游5 Mb处各设计1对在亲本间具有多态性的Indel标记,经聚丙烯酰胺凝胶电泳对两亲本和BC1F5:6的1个家系群体进行基因型鉴定。用Excel 2019对群体基因型结果进行筛选,对照亲本带型,分别获得此群体中每个Indel标记处两亲本基因型个体,结合测得的群体株高表型,计算对应的株高平均值,基于双样本等方差t检验分析不同基因型之间的株高表型差异。
2 结果与分析
2.1 株高表型变异分析
对2016和2017年北京及新疆4个环境的株高数据进行基本的描述统计分析,结果见表1。株高变化范围在154.94~271.63 cm之间,其中17XJ(2017年,新疆)的平均株高明显高于其他3个环境下的平均株高,为222.78 cm;16XJ(2016年,新疆)的变异系数最大,为14.27%。进一步对株高进行方差分析发现,基因型之间存在极显著差异(表2),表明基因型之间的差异可能与遗传因素有关。玉米株高在不同环境中均符合数量性状分布特点,不同环境间均呈显著线性相关,相关性系数在0.55~0.83范围内(图1),其中2017年北京和新疆2个环境存在最强的显著相关性,表明该群体在不同环境下的株高有共同的遗传基础,同时环境条件的改变也会对玉米株高产生一定程度的影响。
2.2 关联分析定位株高相关QTL
本研究对基因型进行过滤筛选后,得到11 781个高质量的多态性SNPs。基于混合线性模型,利用BLUE值和SNP基因型数据进行关联分析,以FDR等于0.05时的P值确定阈值(图2)。从曼哈顿图和 Q-Q 图可以看出,利用16BJ、16XJ、17BJ环境下的株高进行GWAS分析,未找到显著性的SNP位点;利用17XJ环境的株高进行GWAS分析,在第2号染色体上发现9个显著关联的SNPs。利用整合4个环境株高得到的BLUE值进行GWAS分析,发现10个显著关联的SNPs,其中9个SNPs位于第2号染色体上,1个SNP位于第5号染色体上。-log10(P)值最大的SNP位点位于第2号染色体的长臂末端,物理位置为194 690 794 bp(玉米B73参考基因组V3版本),表明该位点附近可能存在一个与株高有关的基因。

表1 BC1F3:4群体在4个环境下株高表型描述性统计Table 1 Descriptive statistics of plant height phenotypes in BC1F3:4 population in four environments
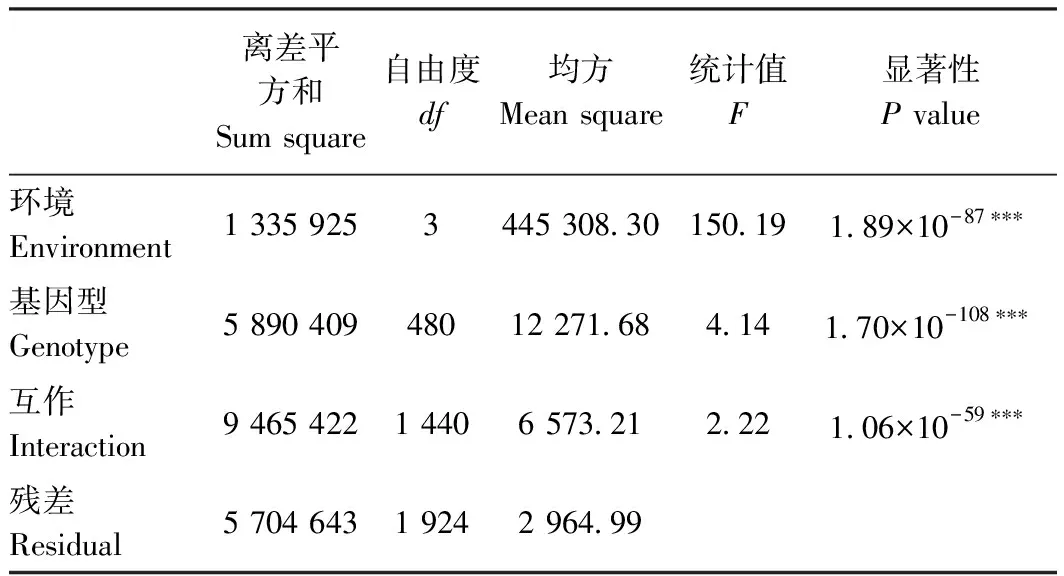
表2 株高的双因素方差分析Table 2 Two-factor variance analysis of plant height
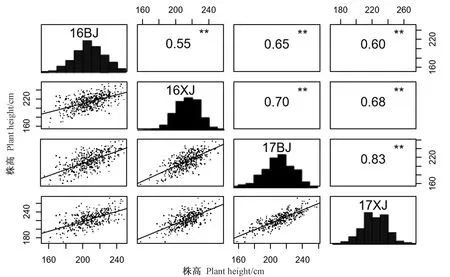
图1 4个环境下株高的分布和相关性分析Fig.1 Distribution and correlation analysis of plant height in four environments

注:A:16BJ株高曼哈顿图和Q-Q图;B:16XJ株高曼哈顿图和Q-Q图;C:17BJ株高曼哈顿图和Q-Q图;D:17XJ株高曼哈顿图和Q-Q图;E:4个环境整合后的曼哈顿图和Q-Q图。Note: A: Manhattan plot and Q-Q plot of plant height in 16BJ. B: Manhattan plot and Q-Q plot of plant height in 16XJ. C: Manhattan plot and Q-Q plot of plant height in 17BJ. D: Manhattan plot and Q-Q plot of plant height in 17XJ. E: Manhattan plot and Q-Q plot of plant height after 4 environment integration.图2 株高性状的关联分析Fig.2 GWAS analysis of plant height
2.3 显著位点表型贡献率及效应分析
为了进一步揭示SNP位点的效应,利用-log10(P)值在显著性阈值4.34以上的SNPs和群体的BLUE值进行线性回归分析,发现表型贡献率在1%以上的显著标记共计2个(图3),分别解释了16%、1%的表型变异,位于第2号染色体的Chr2_194690794标记的表型贡献率明显高于第5号染色体的Chr5_214144001标记。通过对这2个标记的加性效应值计算,发现Chr2_194690794标记具有4.23的正效应,Chr5_214144001的效应值为2.66,PH4CV基因型起正向贡献。
2.4 显著位点基因型验证
依据全基因组关联分析结果,于关联最显著的位点Chr2_194690794两侧5 Mb范围内各设计了1对Indel标记(表3),对两亲本和 BC1F5∶6家系群体进行基因型鉴定,筛选出两亲本基因型个体并计算相应株高表型平均值,通过双样本等方差t检验比较各标记双亲基因型间株高的差异(图4),发现两标记处亲本基因型间株高均在P<0.05水平上存在差异,进一步证实了第2号染色体上存在一个控制玉米株高的QTL。
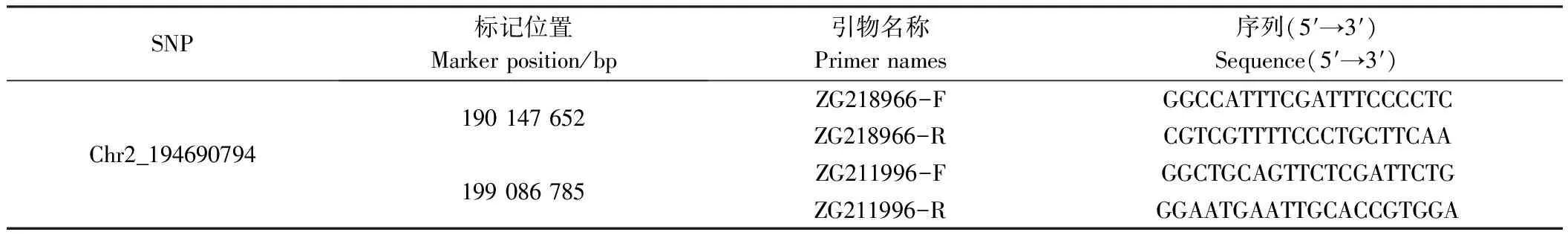
表3 BC1F5:6群体基因型鉴定Indel标记引物信息Table 3 Indel marker primer information for BC1F5:6 population genotyping

注:柱子上的数据为加性效应值。Note: The data on top of each column is additive effect.图3 显著性SNP标记表型变异率Fig.3 Phenotypic variation explained by significant SNP markers
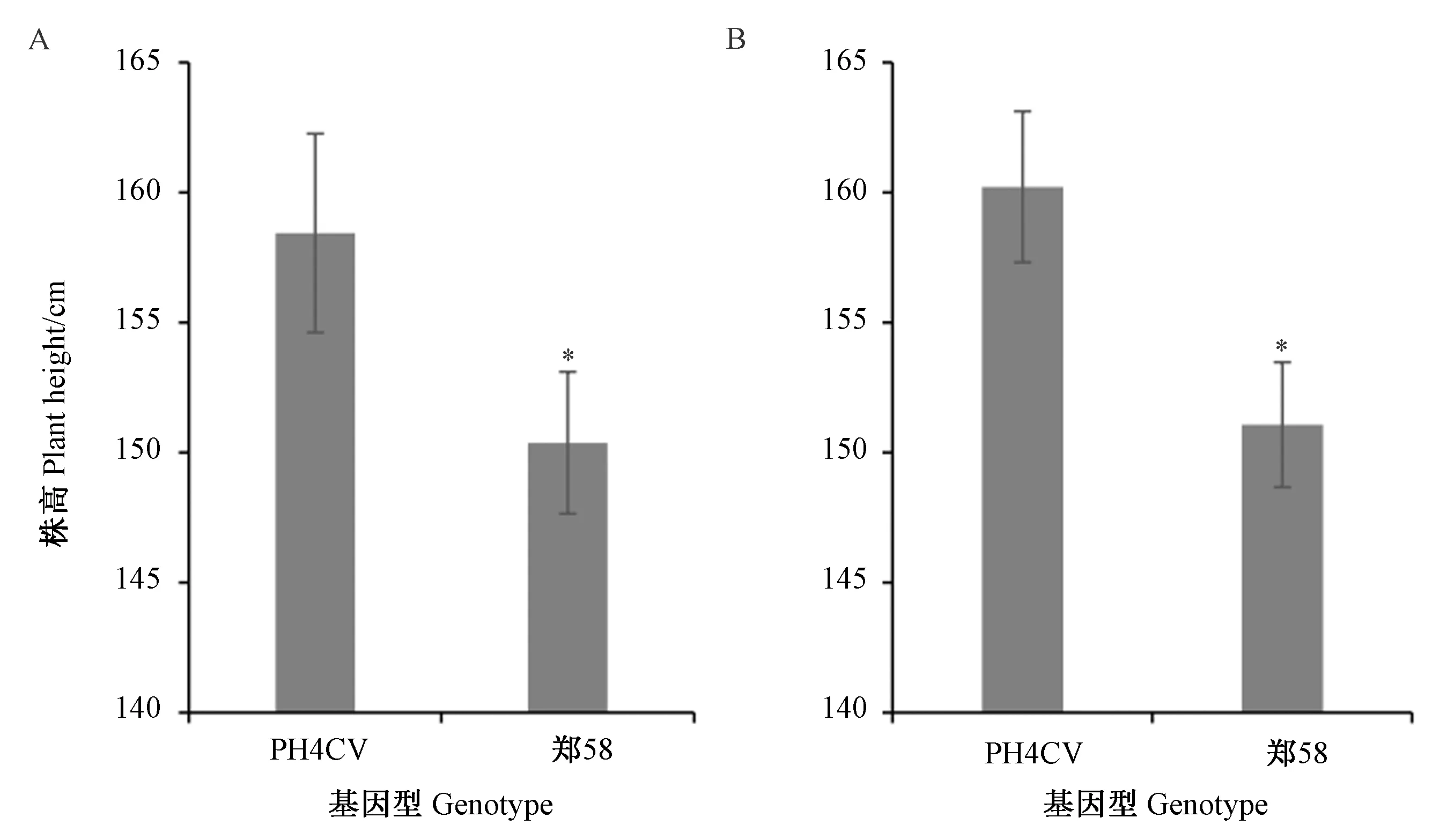
注:*表示在P<0.05水平上差异显著。Note: * indicates significant difference at 0.05 level.图4 BC1F5:6群体显著标记处差异显著性分析Fig.4 Significance analysis of BC1F5:6 population at significant markers
3 讨论
近年来随着对玉米株型性状改良的不断深入研究,研究学者们通过不同研究策略在玉米全基因组的不同染色体上已经鉴定到大量株高QTL[11-13]。本研究以PH4CV和郑58为双亲构建得到包含481个家系的BC1F3∶4群体,利用11 781个高质量SNP标记,通过多年多点的表型及基因型鉴定,进行基于混合线性模型的关联分析发现位于第2号染色体长臂末端的Chr2_194690794标记处存在一个与株高显著关联的位点,该位点所在区域与前人研究鉴定到的株高QTL位置相近。如尤诗婷等[23]使用自交系B73分别与矮杆自交系K22和TY22构建BC2F5群体,基于高密度SNP图谱挖掘株高相关QTL,最终在2个群体中共鉴定到6个QTL,其中2号染色体上的qPH2-2在两群体及两个环境下均可检测到,该位点的表型贡献率达29.55%。于永涛等[24]在其附近也定位到株高相关QTL。李浩川等[25]以郑单958及农大高诱5号为亲本构建的DH系群体为试验材料,利用复合区间作图法在两个地点均检测到位于2号染色体主效QTL,且表型贡献率均达到15%以上。这些结果进一步证明在玉米第2号染色体长臂上存在一个株高主效QTL,同时表明玉米株高可由多个主效基因和微效基因共同控制,印证了株高性状的遗传结构复杂性。
在玉米生产及育种研究中,随着分子标记的广泛应用,研究者们已利用骨干亲本构建群体解析了产量及其他重要性状的遗传基础[26]。如前人利用骨干自交系“黄早四”构建的重组自交系(recombinant inbred line,RIL)群体,对玉米的株高、穗位高等株型性状[27]及产量[28]进行了遗传结构分析,并发掘了相关QTL。但是不同历史时期的主流栽培品种不同,利用育种中已经淘汰的材料进行QTL定位,检测到的QTL并不能反映主流栽培品种中性状的遗传变异基础。本研究所使用的两个优良自交系亲本PH4CV和郑58分别为先玉335和郑单958[29]的亲本。这2个品种属于目前我国广泛种植的主要玉米杂交种,并且在玉米遗传育种中广泛应用。我国目前有许多玉米品种含有这2个杂交种亲本的血缘(maizedata.cn)。利用这2个品种的亲本进行株高等性状的QTL定位更能反映目前主要玉米杂交种的遗传变异情况。因此本研究中的所定位的QTL对于株高的遗传改良具有一定的参考价值。
4 结论
本研究通过全基因组关联分析,于第2号染色体上检测到显著关联的SNP标记,其中-log10(P)值最大的SNP标记为2号染色体的Chr2_194690794。经过进一步表型变异率及效应值分析,发现Chr2_194690794,解释了16%的表型变异,效应值为4.23厘米,且是来源于PH4CV的正效应。对显著位点进行基因型验证的结果表明,Chr2_194690794两侧标记处亲本基因型间玉米株高均存在差异,进一步证明了此位点与玉米株高高度相关。
猜你喜欢
中国糖料(2023年4期)2023-11-01 09:34:46
农村百事通(2019年17期)2019-10-08 02:24:55
中国果业信息(2019年11期)2019-01-05 20:47:24
现代园艺(2017年21期)2018-01-03 06:41:32
现代农业科技(2017年1期)2017-03-06 23:08:18
江苏农业科学(2016年8期)2017-02-15 19:19:29
江苏农业科学(2016年8期)2017-02-15 19:19:15
西南农业学报(2016年5期)2016-05-17 05:42:23
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
医学研究杂志(2015年5期)2015-06-10 06:43:26
