
基于数字基因表达谱筛选双侧去势大鼠附睾的差异表达基因
2022-06-30 08:08赵振军
烟台大学学报(自然科学与工程版) 2022年3期
李 岩,石 慧,赵振军
(烟台大学生命科学学院,山东 烟台 264005)
附睾是高等动物重要的雄性生殖器官,具有多种生理功能,如参与精子成熟和浓缩、分泌和重吸收各种分子和蛋白质等,且具有储存精子的功能[1-4]。附睾发育和正常功能的维持主要受雄激素和睾丸因子的调节。在双侧去势的动物模型中,附睾的重量将减少至正常重量的25%。若给去势动物补充睾酮可使附睾重量增加约三倍,但即使补充超生理剂量的睾酮,附睾重量仍不能完全恢复至去势前重量[5]。 在分子水平上,双侧去势最终会导致附睾基因表达谱的改变。CHAUVIN和GRISWOLD利用Affymetrix芯片平台,在双侧去势小鼠附睾头、体、尾中筛选出50个受双氢睾酮调控且上调两倍以上的转录本,如Adam7、Gstm2和Erabp等[6]。此外,EZER和ROBAIRE构建了成年Brown Norway 雄性大鼠的双侧去势模型,并采用芯片技术检测了474个cDNA在去势组和假手术组大鼠附睾中的表达水平差异,发现去势7 d后Gpx-1、GST、热应激基因及细胞凋亡相关的基因呈现出不同的表达趋势,表明附睾基因表达的调控是极其复杂的[7]。目前为止,双侧去势对Sprague-Dawley(SD)雄性大鼠附睾基因表达水平的影响尚缺乏较为系统的研究。
筛选在不同的组织、发育进程或特定病理过程中差异表达的基因对于基础研究和药物靶标的研究都至关重要。数字基因表达谱技术(DGE)利用新一代高通量测序技术和高性能计算分析技术,能够全面、经济、快速地检测某一物种特定组织在特定状态下的基因表达情况。目前此技术已被广泛应用于基础科学研究、医学研究和药物研发等领域[8]。如SUN等利用DGE和生物信息分析技术研究了低精子活力和高精子活力公鸡睾丸中的全局差异表达基因,为深入了解精子运动的潜在遗传调控提供数据支持[9]。然而采用DGE技术研究双侧去势大鼠附睾差异表达基因在国际上尚未见相关报道。
本研究采用DGE技术在全基因组水平上鉴定了假手术对照组和双侧去势SD雄性大鼠术后第7天附睾中的差异表达基因。已有研究表明,去势后第7天存活的附睾上皮细胞的转录水平变化最具有代表性,因在这一时间节点已检测不到凋亡细胞[10]。随后,我们进行了综合生物信息学分析,以确定基因的表达模式及其在附睾中参与的生物学通路。本研究将为进一步研究附睾中受雄激素及睾丸因子调控的差异表达基因的生物学功能提供数据资源。
1 材料与方法
1.1 实验动物
SPF 级成年雄性SD大鼠(30只,周龄为12周,体重均达到350~400 g)购买于济南朋悦实验动物繁育有限公司,动物许可证号:SCXK(鲁)2019-0003,质量合格证流水号:1107261911003748。实验动物饲养温度为20.0±2.0 ℃,相对湿度为 45%~70%,采用人工照明,且12/12 h 明暗交替的条件环境下适应7 d,期间自由饮食饮水。本研究动物实验经过烟台大学动物伦理委员会审查批准。
SD大鼠随机分为假手术对照组(Con-EP)和双侧去势组(Cas-EP),每组10只。大鼠腹腔注射戊巴比妥钠(30 mg/kg体重)麻醉。然后结扎输精管和睾丸动静脉,从结扎点取出睾丸,将完整的附睾留在阴囊区。对照组同时行假手术。术后恢复7 d后,从去势和假手术对照组大鼠中取出全部附睾立即进行RNA提取,或冷冻于液氮中,-80℃保存以备RNA提取。
1.2 文库构建及测序
大鼠附睾总RNA提取严格按照RNAiso-Plus试剂盒(TaKaRa,D9108A)说明书进行。Agilent 2100评估总RNA的纯度和质量。经检测RIN≥7.0且28S/18S≥1.0,说明提取的RNA足以用于随后的文库构建和测序。RNA样品寄送至深圳华大基因股份有限公司进行文库构建,随后利用Illumina HiSeq 2000系统测序。
1.3 tag注释和基因表达水平量化
将raw tag转换为clean tag需要五个步骤:(1)去除3′端接头以保留21nt长序列;(2)删除空读取(仅包含接头序列但不含tag的序列);(3)去除低质量标签(序列未知的tag);(4)去除过长或过短的tag,仅保留21nt的tag;(5)删除只有一个拷贝数的tag(可能是因为排序错误)。随后将得到的clean tag匹配到不超过1个核苷酸错配的参考序列。对匹配到多个位点的clean tag进行过滤,只保留明确的匹配tag。为了鉴定基因表达,计算每个基因的明确tag数,然后标准化为每百万个标签的转录本数(TPM)。
1.4 差异表达基因的提取
为了挖掘双侧去势后大鼠附睾中差异表达的基因,本研究采用Audic 等提出的改进算法[10]。假设A基因对应的clean tag数为x,每个基因的表达水平仅为所有基因表达水平的一小部分,因此P(x)符合泊松分布。此外,假设样本1和样本2的clean tag数分别为N1和N2,而样本1和样本2中基因A的clean tag数分别为x和y。基因A在两个样本中表达的概率相等,可用公式计算:
利用Benjamini-Hochberg方法[11],通过假发现率(FDR)控制获得的P值。提取FDR≤0.001且log2(倍数改变)≥1的差异表达基因。用皮尔逊相关系数测定了两个库间的相关性。
1.5 Gene Ontology(GO)及KEGG Pathway显著性富集分析
GO功能显著性富集分析用于分析所有匹配基因以注释它们潜在的生物学功能。Pathway分析利用KEGG生物通路数据库进行富集度分析。对筛选出的差异表达基因经KEGG数据库注释,可准确定位到对应的Pathway条目中,经超几何检验,得到差异基因显著富集通路。P值用Bonferroni校正法(Q值)校正,并将阈值设置为0.05以表示显著富集的标签。
1.6 差异表达基因的实时定量PCR分析
采用实时定量PCR(real time quantitative PCR,qPCR)检测相关差异基因的表达水平。首先取1 μg总RNA作为反转录模板,利用ReverTra Ace试剂盒(日本TOYOBO公司,FSK-100)合成cDNA第一条链。然后在QIAGEN的Roter Gene Q仪器上使用Platinum SYBR Green qPCR SuperMix-UDG试剂盒(Life Technologies,11733-038)进行qPCR扩增。qPCR反应体系共50 μL:cDNA 1μL,SYBR green 25 μL,正向引物(0.3 μmol/L) 1.5 μL,反向引物(0.3 μmol/L) 1.5 μL,灭菌H2O 21 μL。反应程序采用两步法扩增:50 ℃ 2 min,95 ℃ 2 min,[95 ℃ 15 s,60 ℃ 30 s] 40个循环。以GAPDH为内源性参照物。2-ΔΔCt方法用于计算受检样本中基因表达水平的差异[12]。所有实验重复三次,引物序列见表1。

表1 qPCR引物序列
2 结果与分析
2.1 测序数据质量评估
为初步筛选假手术对照组和双侧去势大鼠附睾中的差异表达基因,本研究分别构建了Con-EP和Cas-EP文库,并用Illumina HiSeq 2000系统测序。在每个库中获得了超过450万个raw tag。在去除低质量的tag之后,约剩余96%的raw tag,即9 143 758个clean tag。其中两个库中共有209 405个不同的clean tag(表2)。

表2 Con-EP和Cas-EP文库测序数据统计
为了评估DGE数据的正态性,我们比较了tag表达的分布,因为tag的拷贝数实际反映了mRNA表达水平[13]。如图1所示,在两个DGE库中,total clean tag和distinct clean tag显示出相似的分布模式,表明在Con-EP和Cas-EP库的构建过程中不存在偏差。在total clean tag中,占优势的高表达tag是那些拷贝数超过100的tag,而在distinct clean tag中,占优势的高表达tag的拷贝数小于5。该结果表明小部分mRNA 高表达,而大部分mRNA低水平表达,符合细胞 mRNA的表达特征。
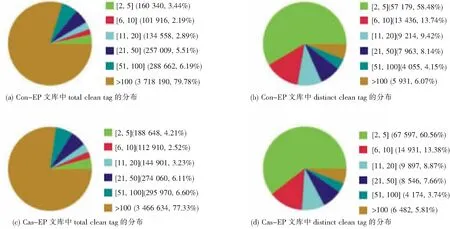
图1 Con-EP和Cas-EP文库中clean tag的拷贝数分布
2.2 大鼠参考基因的标记定位
为了将DGE图谱转化为基因表达,将两个DGE文库的tag序列与NCBI注册的大鼠基因数据库进行比对。Con-EP和Cas-EP文库中匹配的clean tag分别为293万和315万,分别占clean tag总数的62.89%和70.32%(表2)。在两个文库中,分别有41164个和46270个匹配的distinct clean tag,分别占42.10%和41.45%。饱和度分析结果表明,当clean tag数量达到200万时,基因数的增长曲线变得平坦,表明两个样本的测序均趋于饱和(图2)。用无歧义标记计算的每百万个标记的转录本数(TPM,transcript per million tags)是表征基因表达水平的一个很好的指标。基因表达的累积分布表明,约有一半的基因表达在10个拷贝以下(log10(TPM)<1),90%的基因表达量不超过100个拷贝,只有一小部分基因高表达(图3)。
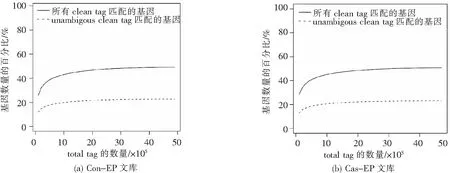
图2 测序饱和度分析
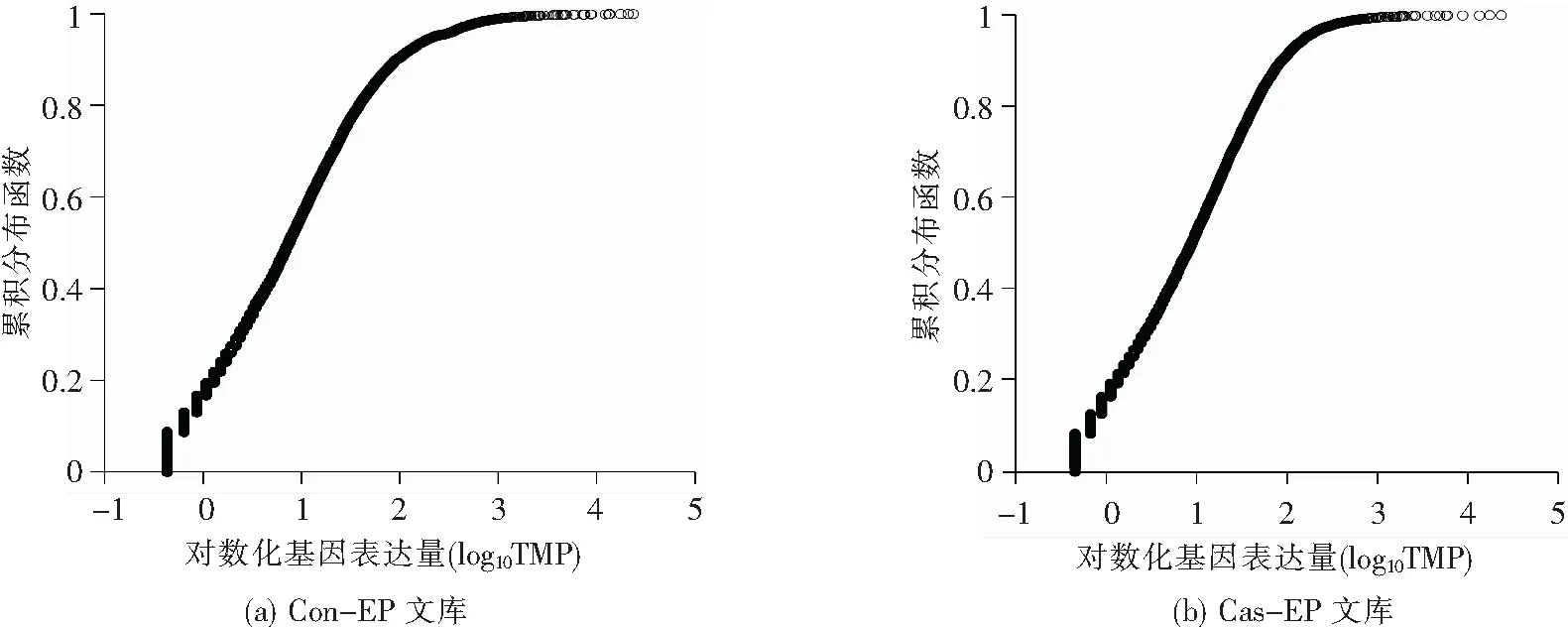
图3 基于unambiguous tag的基因表达累积分布
2.3 双侧去势大鼠附睾差异表达基因分析
为了比较双侧去势大鼠附睾差异表达基因,将tag的基因表达水平在Con-EP和Cas-EP两个文库中的分布进行了标准化,并提取FDR≤0.001 且 log2fold-change≥1的显著差异表达的tag。两个文库之间共有11 191个tag(匹配到2 448个基因)发生了显著变化。其中,1 632个基因在双侧去势后的大鼠附睾中表达上调,816个基因表达下调。基因表达水平的倍数变化(log2)介于-16.5~10.4之间。约90%以上的基因(2 274)上调或下调1.0到5.0倍之间(图4(a))。两个文库的皮尔逊相关系数仅为0.658,表明去势对大鼠附睾基因表达谱有显著影响(图4(b))。

图4 Con-EP与Cas-EP文库差异表达基因分析
为验证DGE技术鉴定的差异表达基因的可靠性,随机挑选6个差异表达的基因(上调基因:Rab3b、Hsd3b6和Hormad2,下调基因Lcn9、Ly6g5c和Smcp)进行qPCR实验。如表3所示,qPCR结果与测序数据一致,证实了测序结果的真实性(P值均小于0.01)。

表3 差异表达基因的qPCR验证
GO富集分析结果表明,所有上调和下调节基因的基因功能分布相似,但上调组突触部分和辅助转运蛋白是唯一的,而电子载体和病毒粒子部分则仅存在于下调组(图5)。
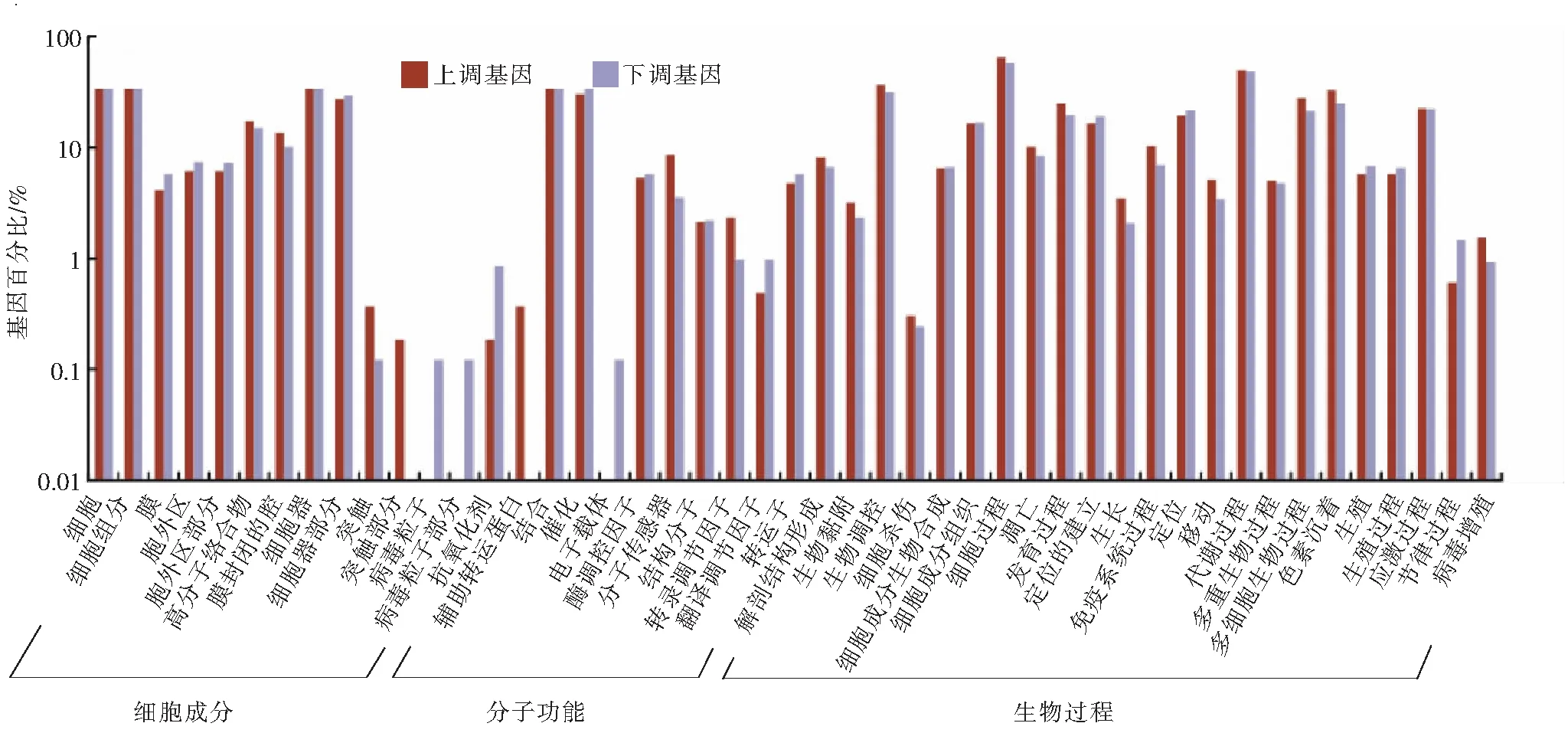
图5 差异表达基因的GO富集分析
2.4 差异表达基因的通路分析
为进一步了解双侧去势后大鼠附睾中差异表达基因的生物学功能,本研究采用KEGG本体论方法对差异基因的功能注释进行分类。在KEGG数据库的2 448个差异表达基因中,有1 881个基因被定位到250条通路中。通路富集分析表明,代谢通路、氧化磷酸化通路以及溶酶体通路是前三条显著富集的途径。
3 讨 论
高通量转录组测序和数字基因表达标签分析对于全局转录组研究十分高效且经济[14-15],利用这些技术可以方便、准确地分析多个样本中差异表达的基因。因此本研究采用数字基因表达谱技术,研究了成年大鼠和双侧去势大鼠附睾中差异表达基因。
数据分析表明,共有2 448个基因在双侧去势大鼠附睾中的表达差异显著,其中1 632个基因上调,816个基因下调。在这些差异表达的基因中,有1 881个基因被定位到KEGG数据库中的250条通路。通路富集分析表明,代谢通路、氧化磷酸化通路和溶酶体通路是三条显著富集的途径。据报道,雄激素状态将显著影响哺乳动物附睾的正常结构和生物学功能的维持,这种对雄激素的依赖性反映在对器官的代谢调控上[16]。在四种主要的附睾上皮细胞类型,即主细胞、基底细胞、狭窄细胞和亮细胞中,主细胞对循环雄激素的剥夺尤为敏感,而其他三种细胞类型受影响较小[9]。双侧去势动物附睾最明显的形态学变化之一是主细胞顶端微绒毛数量减少。雄激素剥夺后,粗糙内质网的数量减少,但高尔基体或线粒体的变化较小[16]。 除影响附睾细胞形态外,雄激素剥夺可能影响实验动物的整体代谢过程,如大鼠体内雄激素对合成代谢反应和为这种反应提供前体或辅助因子的其他途径有相当大的影响。因此,雄激素缺乏的动物附睾脂质合成减少,戊糖磷酸途径和丙酮酸羧化酶的活性也减少。另一方面,在成年动物中,雄激素剥夺后细胞DNA和RNA显著减少。在蛋白质代谢方面,雄激素对蛋白质合成速率和蛋白质合成类型影响不大,但对某些特定的分泌性蛋白质影响较为显著。然而,雄激素可以增加附睾蛋白质的平均周转时间[16]。
本研究采用qPCR方法验证了随机选择的6个上调或下调表达的差异基因。结果表明,这些基因在去势组和假手术对照组附睾中的表达趋势与DGE测序丰度的变化一致,从而证实了DGE测序技术的可靠性。值得注意的是,在去势后表达显著下调的基因Lcn9属于脂质运载蛋白(lipocalin)家族。该家族是一个结构保守、功能多样的蛋白质家族,广泛参与了免疫反应、细胞生长和代谢调节、铁转运和前列腺素合成等,因此在附睾中具有重要的生物学功能[17]。已有文献报道,大鼠Lcn9 mRNA仅在附睾头段呈特异性表达,且在双侧去势大鼠的附睾中,Lcn9 mRNA水平在去势后1 d显著下降,在随后的3~7 d基本检测不到其表达,即使给去势7 d后的大鼠补充睾酮,仍未能检测到。此外,免疫组化结果表明,Lcn9蛋白在45日龄大鼠附睾头段/起始段区域上皮细胞中开始表达,此后其表达逐渐增加并保持在高水平,表明Lcn9蛋白参与了附睾的发育和成熟过程[18]。除Lcn9外,DGE测序中还检测到Lcn家族的其他成员在去势后的附睾中表达显著下调,如Lcn2、Lcn5、Lcn6、Lcn8、Lcn10、Lcn12和Lcn13,它们基因表达水平下调倍数(log2)分别为1.3倍、4倍、3倍、8倍、10倍、6倍、4倍。因此Lcn基因家族为DGE筛选出的受雄激素及睾丸因子调控的重要候选基因家族,其在附睾中的生物学功能及调控精子成熟的机制有待于深入研究探讨。
此外,本科研组前期研究筛选出59个去势前后差异表达的microRNA,其中24个上调,35个下调,且在不同周龄的大鼠附睾中呈现出发育时序规律性变化,表明它们是受雄激素水平调节的[19]。本研究中去势引起的雄激素水平的变化可能导致某些microRNA表达水平的变化,而附睾基因表达谱变化可能是这些microRNA精细调控的结果。建立microRNA与本研究筛选出的差异表达基因的对应关系将成为今后研究工作的重点。
综上,本研究是对双侧去势大鼠附睾基因表达谱的首次系统性研究,将为进一步研究附睾中受雄激素及睾丸因子调控的差异表达基因的生物学功能提供数据资源。
猜你喜欢
保健与生活(2022年12期)2022-06-09
体育科技文献通报(2022年3期)2022-05-23
现代仪器与医疗(2021年6期)2022-01-18
孔学堂(2020年1期)2020-06-01
中华诗词(2019年1期)2019-11-14
养生保健指南(2017年12期)2017-12-07
中外医疗(2016年33期)2017-03-02
环球人物(2015年12期)2015-09-10
中国医药科学(2014年24期)2015-03-16
读书(1980年9期)1980-07-15
