
石墨炔零价金属原子催化剂的合成及应用
2022-06-29 12:35:12陈朝阳薛玉瑞李玉良
高等学校化学学报 2022年5期
陈朝阳,薛玉瑞,李玉良,2
(1.山东大学物质创制与能量转换科学研究中心,化学与化工学院,前沿交叉科学研究院,济南 250100;2.中国科学院化学研究所有机固体实验室,北京 100190)
原子催化剂因具有独特的电子结构、高催化活性和高选择性等优异性能,被认为是理想的催化模型体系[1~6].原子催化剂每一个独立的金属原子都完全暴露,作为反应活性位点与电解液充分接触,可以实现最大的金属原子利用效率.由于这些独立的活性位点的同质性,原子催化剂能将均相催化剂和异相催化剂的优势充分融合,为真正从原子尺度上理解催化反应机理提供了契机,已成为催化和能源等领域的研究前沿.科学家们已在单原子催化的基础和应用方面进行了许多研究[7~39].近年来,人们通过不同合成手段将过渡金属原子(Fe[40~42],Co[43,44],Ni[45,46]等)和贵金属原子(Pt[47,48],Pd[49,50],Rh[51]等)锚定在g-C3N4[52]、有机金属框架化合物[53]、有机共价框架化合物[54]、石墨烯[55]和MoS2[56]等不同的基底上.然而,金属原子易迁移聚集一直是合成高效单原子催化剂所面临的重要挑战.科学家们通过降低金属原子负载量、调控金属-载体相互作用以及在基底上引入空穴或缺陷等方法[57~61]在一定程度上抑制了单原子催化剂的自聚问题.但是受限于严格的合成条件、困难复杂的制备工艺、特定的仪器要求、严格的质量控制和低生长率等因素,传统的单原子催化剂在提高金属原子稳定性、合成具有明确价态和结构组成的原子催化剂、精确控制催化过程中的化学结构和电荷转移等方面缺乏突破,成为限制金属原子催化剂实际应用的巨大障碍[62].因此,如何成功地将单个金属原子锚定在合适的载体上,实现原子催化剂的合成和真正应用仍然是一个重大挑战.
石墨炔具有我国自主知识产权,是我国科学家在国际上引领的研究领域,其独特的原位生长特性、丰富的化学键、高共轭的大π键和天然的孔洞结构以及在碳材料中独有的“炔-烯互变”性质,及其在电子转移、电荷传输、离子输运、能量传递与转换等方面表现出的优越性质,激发了研究人员的新思维和新理念.石墨炔的sp和sp2杂化使其表面电荷分布极不均匀,这一传统碳材料不具备的特点赋予了其无限的天然活性位点和高本征活性,使其在结构和性质方面展示出巨大的优势和先进性.2010年,李玉良等[63]首次利用化学合成方法大面积制备出石墨炔薄膜以来,石墨炔在光电催化、能量存储、生物检测、光电探测、电化学驱动器和生命科学等领域展示出无限的潜力,并获得了许多基础和应用上的原创性研究成果[3,5,6,64~80].在中国科学院科技战略咨询研究院、中国科学院文献情报中心与科睿唯安联合面向全球发布的《2020研究前沿》报告中,“石墨炔研究”同时入选化学与材料科学领域“Top10”热点前沿.
与传统的碳材料结构不同,除了碳碳双键之外,石墨炔还具有sp/sp2共杂化的独特性质,使π/π*能朝着垂直于碳碳三键的任何方向旋转,从而更易于和周围的金属原子相互作用,实现金属原子在石墨炔上的稳定锚定.得益于这些优点,石墨炔为人们提供了合成结构明确、性能优异的的原子催化剂的新途径.2018 年,Li 等[3]利用石墨炔丰富的炔键、孔洞结构及其与金属原子之间的相互作用,并结合多孔结构的空间尺度效应,首次提出合成零价过渡金属原子的新理念,首次成功锚定零价过渡金属原子(图1)[3],克服了传统单原子催化剂易迁移、聚集、电荷转移不稳定等问题,真正实现了零价金属原子催化,为发展新型高效催化剂开拓了新的方向,并将该零价金属原子催化剂命名为“原子催化剂”.石墨炔原子催化剂展现出高稳定性、高选择性、高活性和高法拉第效率等性能,并被率先应用于常温常压下固氮制氨、分解水制氢等的重要领域.本综述从石墨炔原子催化剂的结构、催化剂的设计、可控合成和结构表征等方面出发,总结了近年来石墨炔原子催化剂在催化和能量转换等领域的研究成果.基于石墨炔基原子催化剂结构与本征性能之间的构效关系,探讨了石墨炔原子催化剂在高性能催化及能量转换领域的应用和发展前景,为实现新概念高性能催化材料的设计合成提供了重要研究思路.

Fig.1 Synthesis and structure diagram of graphdiyne atomic catalysts[3]
1 石墨炔零价过渡金属原子催化剂的合成与表征
科学家们一直期待着零价过渡金属原子催化剂的出现.然而受限于复杂和苛刻的合成条件,已报道的单金属原子催化剂存在价态不确定(正价态或混合态)和化学结构不明确等缺点,制备高效、稳定的零价过渡金属原子催化剂是催化领域一个巨大的挑战.目前,合成石墨炔零价金属原子催化剂的方法主要为电沉积法[3,73,77,80]、水热法[5]、湿化学法[79]以及金属离子在石墨炔上自还原[76]等.

Fig.2 HAADF⁃STEM images of Ni/GD and Fe/GD(A—L),element distribution diagram of Ni/GD(M),off site EXAFS spectra of Ni and Ni foil at Ni K⁃edge(N),element distribution diagram of Fe/GD(O),off site EXAFS spectra of Fe and Fe foil at Fe K⁃edge(P)[3]
利用石墨炔丰富的炔键、孔洞结构及其与金属原子之间的相互作用,并结合多孔结构的空间尺度效应,李玉良院士团队[3]首次成功实现了零价过渡金属原子的锚定[图2(A)],获得了零价金属原子催化剂,是催化领域的重大突破.如,在常温常压下,利用简单的原位电化学沉积一步法,即在石墨炔表面成功锚定了单个金属原子镍(Ni)和铁(Fe),成功制得了石墨炔基零价Ni和Fe原子催化剂(Ni0/GDY,Fe0/GDY).球差校正高角环形暗场扫描透射成像技术(HAADF-STEM)和元素面扫描成像结果表明,金属Ni和Fe(图中亮点代表金属原子)分别是以单原子形式相互独立地均匀锚定于石墨炔表面;近i边X射线吸收精细结构谱(EXAFS)结果表明,样品中无金属-金属键的存在,进一步证明单个金属Ni和Fe原子催化剂的成功制备.通过对HAADF照片的统计分析,可以得到Ni0/GDY和Fe0/GDY中金属原子具有非常窄的粒径分布,Ni 为(0.123±0.040)nm,Fe为(0.102±0.033)nm,再次验证了Ni0和Fe0原子在GDY基底上的优异分散.
近边X射线吸收精细结构谱(XANES)能够非常灵敏高效地测试到同一元素的价态变化,可以作为辨别同一种元素不同价态的指纹谱.因此,李玉良院士团队[3]对所有的样品进行了同步辐射原位测试.为了精确地证明金属原子的价态,他们利用纯的镍箔(铁箔)作为对照样品.如图3(A)和(B)所示,Ni0/GDY 和Fe0/GDY 结合能与零价金属的结合能一致,充分证明两种催化剂中Ni 和Fe 都为零价.他们进一步对Ni0/GDY和Fe0/GDY结构稳定性进行了表征.XPS分析显示,两种催化剂的sp/sp2比值都为2,说明金属原子锚定过程中没有破坏石墨炔结构.拉曼图谱显示,相比于纯石墨炔(0.77),Ni0/GDY 和Fe0/GDY 中D 带和G 带的强度之比(Ni0/GDY:0.87,Fe0/GDY:0.85)显著增强,证明石墨炔基原子催化剂具有更多的活性位点,有利于催化活性的提升.为了进一步考察石墨炔基零价金属原子催化剂的的稳定性,他们对新制备的样品分别在反应前后甚至是在高温条件下处理后的样品进行了XANES和EXAFS表征,结果显示,锚定于石墨炔表面的金属原子的价态以及锚定位置仍未发生任何变化.这些结果都充分证明了零价金属原子在石墨炔表面的高效稳定锚定.

Fig.3 EXAFS(A) and XANES(B) spectra of Ni/GD and Ni foil at Ni K⁃edge(A,B),EXAFS(C) and XANES(D)spectra of Fe/GD and Fe foil at Fe K⁃edge,XAS spectra of Ni/GD at 5%H2/He and different temperatures(Ni foil as a control)(E,F),XAS spectra of Fe/GD at 5% H2/He and different temperatures(Fe foil as a control)(G,H)[3]
他们还进一步利用理论计算的方法,从键合能量角度对石墨炔基零价金属原子催化剂中金属原子的价态以及金属原子和石墨炔之间的相互作用进行了研究.如图4(A)和(B)所示,通过对石墨炔与金属原子之间的相互作用强度进行计算,确定金属原子稳定锚定于石墨炔中炔环的顶端相邻炔键中间位置(A1).图4(C)中理论模拟结果显示,Ni2+样品则表现出非常显著的开壳效应,零价的fcc-Ni表现为典型的闭壳效应[图4(D)],且仅有非常轻微的3d-3d轨道重叠.而与零价的fcc-Ni结果一致,Ni0/GDY样品则表现出非常明显的闭壳效应[图4(E)],说明Ni0/GDY中的金属为零价态.此外,理论计算得到的Ni—C 键长(0.1753 nm)与Ni3d轨道能量(9.79 eV)均与实验测量结果一致.实验和理论计算结果都充分证明了零价金属原子在石墨炔上能够稳定存在,即已成功制备了零价金属原子催化剂.石墨炔原子催化剂中金属原子与石墨炔之间独特的不完全电荷转移实现了金属原子的高效稳定锚定,产生更多活性位点数量,提高了其电荷转移能力,赋予了催化剂较高的催化活性.

Fig.4 Possible adsorption sites of Ni/Fe atoms along route A and route B in GD layer(top:top view;bottom:side view)(A),binding energy of Ni/Fe atoms adsorbed by double⁃layer GD vs.the displacement along the A and B directions(B),3D orbital binding energy of the anchored Ni position determined by the open shell charge overlap and the closed shell charge overlap[NiO(C),Ni on GD(D),and Ni-fcc(E)],and change of orbital energy related to the newly formed Ni-C(F)[3]
该石墨炔基零价金属原子催化剂制备方法具有很强的普适性,可用于锚定多种零价金属原子,通过该方法首次实现了Ni0/GDY,Fe0/GDY,Cu0/GDY,Mo0/GDY,Co0/GDY 和Pd0/GDY 零价金属原子催化剂的制备.如,Yu 等[73]通过电沉积的方法合成了零价钯原子催化剂(Pd0/GDY),扫描电子显微镜(SEM)和透射电子显微镜(TEM)结果表明,反应过程中没有Pd纳米颗粒的形成[图5(A)~(H)],元素面扫结果显示,Pd原子均匀分布于石墨炔表面[图5(M)~(P)].为了更进一步观察制备的Pd/GDY中金属原子存在状态,他们首先对其进行了HAADF-STEM表征[图5(I)~(L)],图中的亮点代表了单个独立存在的Pd金属原子,证明了Pd在石墨炔基底上的高度分散分布.通过对HAADF照片的统计分析,可以得到Pd0/GDY中金属原子具有非常窄的粒径分布(0.36±0.01 nm).此外,他们对所制备的样品结构进行了表征(图6).如图6(A)所示,EXAFS测试结果显示,Pd0/GDY样品的主峰在0.15 nm左右,明显小于Pd 箔中Pd—Pd 键长(0.25 nm),进一步证明Pd/GDY 中Pd 以单分散的原子存在.XANES 结果显示,Pd/GDY 的主峰和金属钯箔的峰相近[图6(B)],证明锚定在GDY 上的Pd 单原子的价态为零价.XRD测试结果进一步证明,所制备的Pd0/GDY样品中无钯金属颗粒的生成.拉曼光谱[图6(C)]和XPS[图6(D)~(F)]测试结果均显示,金属原子锚定过程不对石墨炔结构产生任何破坏.Pd3d的XPS 谱图[图6(F)]分析结果进一步确认了锚定于石墨炔表面的Pd为零价态.DFT研究发现,不同数目的Pd原子锚定在GDY上的Pd的4d轨道的相对特征[图6(G)],两种锚定方式下Pd都展现了闭壳效应[图6(H)和(I)],这说明Pd原子与相邻的C原子之间有着强烈的轨道重叠(p-d耦合).在三重锚定的情况下,相邻的Pd原子之间存在着d-d轨道共振耦合,从而增强了电负性.图6(J)展示了Pd4d轨道和Ni3d轨道的偏电子态密度投影(PDOS),结果显示,Pd4d轨道有着更深的4d带中心,意味着更多的4d电子与相邻C原子的p轨道进行耦合.Pd的4d轨道与相邻C的p轨道重叠的价电子定域轨道图像证明,锚定在石墨炔上的Pd原子与石墨炔本体之间存在着独特的不完全电荷转移.这也是金属原子能在石墨炔上高效稳定锚定和产生优异催化性能的关键.

Fig.5 SEM images of Pd0/GDY(A,B),TEM(C) and high resolution TEM(D) images of Pd0/GDY,HAADF images of different regions of Pd0/GDY nanosheets(E—H),enlarged images of the selected area in (E)—(H)(I—L),STEM⁃HAADF images of Pd0/GDY nanosheets and corre⁃sponding element distribution diagram of Pd and C atoms(M—P)[73]
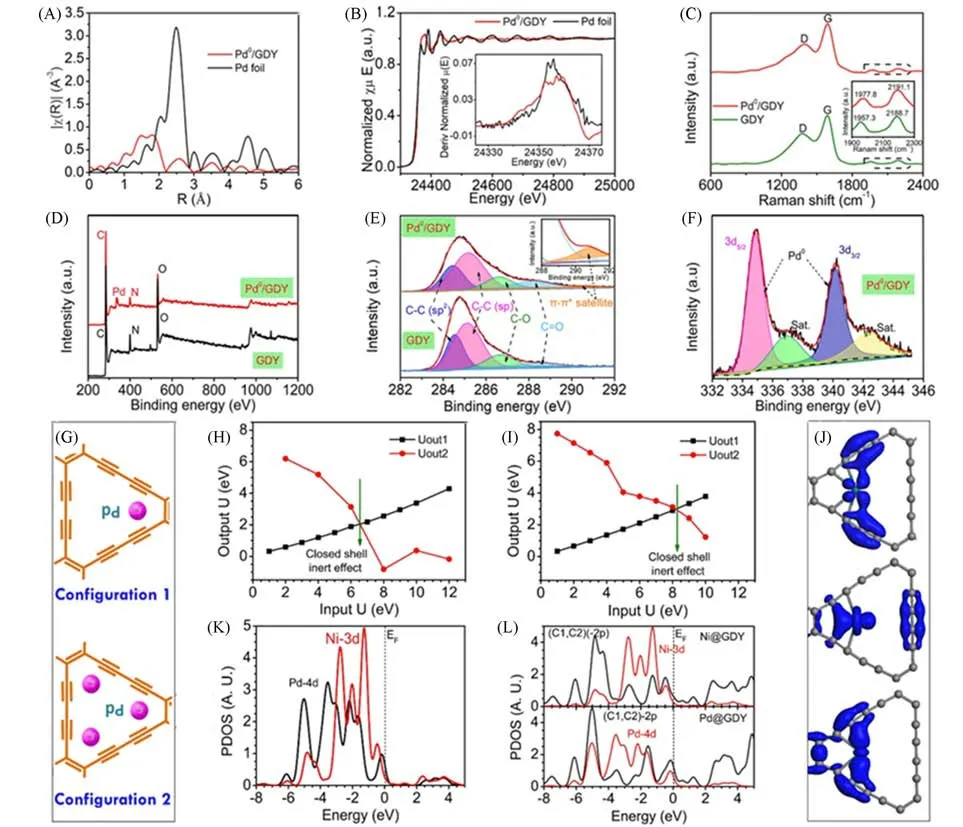
Fig.6 EXAFS spectra of Pd0/GDY and Pd foils in Pd K⁃band(A),normalized spectra of Pd0/GDY and Pd foil in Pd K⁃band(inset:first derivative curve)(B),Raman spectra of Pd0/GDY and initial GDY(inset:signal of alkyne bond structure on GDY skeleton)(C),XPS spectra of Pd0/GDY and initial GDY(D),C1s high resolution XPS spectra of Pd0/GDY and initial GDY(inset:enlarged image of 290.8 eV region)(E),XPS spectra of Pd3dof Pd0/GDY(F),in GD,the single(high) and triple(low) anchor positioning points(position a) of Pd(G),orbital potential energy projection(Uout1 and Uout2)of Pd⁃4d in single(H)and three anchor positioning points(I),PDOS compari⁃son of Pd⁃4d and Ni⁃4d bands in GD⁃Pd and GD⁃Ni(J),compared with Ni⁃4d and (C1,C2)⁃2p bands,the total contribution of Pd⁃4d and (C1,C2)⁃2p bands to PDOS(K),real space 3D orbit profile of three main p⁃d band overlapping peaks observed from PDOS between Pd⁃4d and(C1,C2)⁃2p band(L)[73]
钼(Mo)是人体及动植物所必需的第二过渡系元素,零价钼(Mo0)已被证明是许多重要化学反应的核心组成部分.然而自然界中Mo普遍以高氧化态化合物形式稳定存在.传统单原子催化剂的制备方法无法得到零价钼原子催化剂.利用石墨炔的独特结构和性质,Li等[5]从理念上创新,提出了锚定零价钼原子的新策略,成功在石墨炔表面负载了的零价钼原子[负载量(质量分数)高达7.5%],并实现了其表面活性组分的高度分散,构筑了双功能石墨炔基零价钼原子催化剂(Mo0/GDY),实验表征结果都证明零价钼原子相互独立、高度分散地锚定于石墨炔表面.值得一提的是,他们首次从实验上获得了单个金属原子在石墨炔上锚定位置的清晰照片(图7).实验结果也进一步证明了理论计算研究模型的正确性.
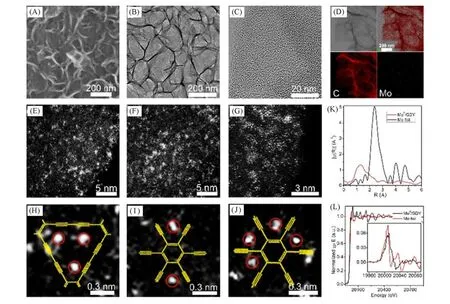
Fig.7 SEM(A),TEM(B) and HRTEM(C) images of Mo0/GDY,STEM image of Mo0/GDY and corresponding element distribution image(D),HAADF⁃STEM images of newly prepared Mo0/GDY samples(E—G),different configurations of each Mo atom fixed on the GDY structure(H—J),EXAFS spectra of Mo K⁃edge of Mo0/GDY(red line) and Mo foil(black line)(K),normalized Mo K⁃edge XANES spectra of Mo0/GDY and Mo foil(inset:calculated first derivative curve)(L)[5]
2 石墨炔零价金属原子催化剂的应用
石墨炔零价原子催化剂的成功合成,解决了传统载体上作为单个金属原子易迁移、聚集和电荷转移不稳定等关键问题.金属原子在石墨炔表面的成功锚定,同时能够显著增加体系的导电性和反应活性位点数量,有利于增强电催化活性.更重要的是,对于石墨炔零价原子催化剂,锚定的零价金属原子能够进一步活化锚定位点附近的新活性位点[3,5,73~75].石墨炔零价金属原子催化剂因其独特的优势,在催化、能源转换等领域展现出了独特优势和无限的潜力.
2.1 高效固氮制氨反应
氨气(NH3)是一种理想的氢能源载体,在储氢领域具有重要地位,同时也是现代工业和农业生产最为基础的化工原料之一,对人类的生产、生活等方面至关重要.然而,目前工业上主要在高温、高压(400~600 ℃和20~40 MPa)等苛刻的条件下合成氨,不仅耗能巨大,更会导致环境的严重污染.因此,如何实现常温、常压下高效合成氨受到科学界和工业界的广泛关注.电化学还原氮合成氨(ECNRR)是一种可以实现常温常压下合成氨的新技术,然而实现该技术的核心是如何获得高活性和选择性的催化剂.人们一直致力于开发新型产氨催化剂(如贵金属和金属氧化物等),但它们较低的产氨效率和低的法拉第效率,以及欠佳的稳定性限制了其用于大规模生产.
近来,我们[5]提出了一种简便的、可规模化的水热原位还原法锚定零价钼原子的新策略,成功在石墨炔表面负载了的零价钼原子(负载量高达7.5%),并实现了其表面活性组分的高度分散.实验表征和理论计算结果均充分证实,Mo0/GDY具有明确的化学结构和确定的化学价态(零价).Mo0/GDY也是第一个能够在常温、常压下高选择性、高活性和高稳定性电催化固氮制氨的零价原子催化剂.如图8 所示,在N2饱和的0.1 mol/L Na2SO4中,Mo0/GDY 在1.2 V 时可达到最大的NH3产率(113.4~145.4)和法拉第效率(15.2%~21.0%),而在酸性条件中(0.1 mol/L HCl),Mo0/GDY 仅需要−0.1 V 的电势即可达到2.0的NH3产率和15.6%的法拉第效率.反应过程中未检测到可能的副产物(肼)的生成,证实了Mo0/GDY催化氮还原具有高度选择性.通过测试比较发现,反应前后Mo0/GDY 的化学结构和价态均未发生变化,并且也与已报道的其它几种石墨炔基金属原子催化剂类似,在酸性条件中Mo0/GDY的优异HER性能也得到了理论和实验上的验证,其仅需48 mV的过电位即可达到10 mA/cm2的电流密度,0.2 V 过电位下的质量活性高达15.10 A/mgmetal,为20%Pt/C 的2.6 倍,这与预测的氢吸附自由能结果一致.
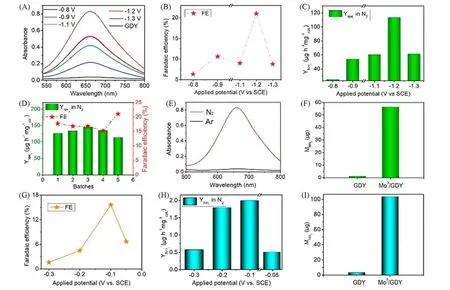
Fig.8 ECNRR performance of Mo0/GDY[5]
Yu 等[76]合成了锚定在GDY 上的零价Pd 原子催化剂,得到了负载量为2.9 μgmetal/cm2的催化剂(图9),经过测试发现,其在中性条件下(0.1 mol/L Na2SO4)具有超高选择性和稳定性,氨气产率达到(4.45±0.30),法拉第效率达到了(31.62±1.06)%,该氨产率超过了目前报道的所有在温和环境下电催化氮气制氨的催化剂,创造了固氮制氨新纪录.此外,他们还测试了其在酸性环境(0.1 mol/L HCl)下的催化性能,发现同样具有高的选择性和催化活性,这为开发高效电催化制氨催化剂提供了非常好的参考.
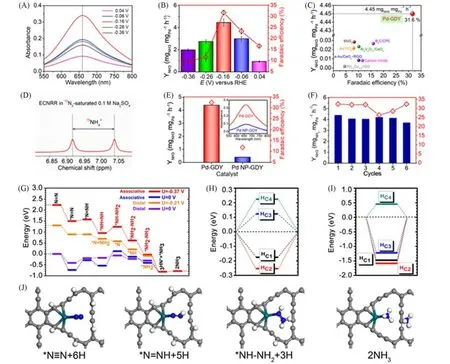
Fig.9 UV⁃Vis absorption spectra of 0.1 mol/LNa2SO4 electrolyte at different potentials for 2 h after ECNRR(A),ammonia yield and Faraday efficiency at applied potential in 0.1 mol/L Na2SO4(B),comparison of ECNRR performance of Pd⁃GDY with other catalysts(C),1H⁃15N NMR spectra of 0.1 mol/L Na2SO4 after ECNRR under 15N2 with Pd⁃GDY as catalyst(D),am⁃monia yield,Faraday efficiency and corresponding UV⁃Vis absorption spectra of Pd⁃GDY and Pd⁃NP/GDY catalysts at −0.40 V after electrolysis for 2 h at room temperature(illustration)(E),stability test of Pd⁃GDY under the following conditions⁃under ambient conditions(the voltage:0.4 V in 0.1 mol/L Na2SO4)(F),ECNRR kinetic steps of Pd⁃GDY(G),H formation energy of Pd⁃GDY on the C side(H),H chemisorption energy of Pd⁃GDY on the C side(I),and structural evolution of ECNRR catalytic process(J)[76]
理论计算结果显示,电催化固氮制氨的决速步主要是与石墨炔(C1,C2)位点相连的不对称氢-脱附[图9(A)].通过比较氢-吸附能,发现C1 和C2 是最有利的氢-吸附能位点.此外,H-化学吸附能表明,C1 和C2 为H-吸附提供了两个最佳活性位点,以实现高效的质子-电子电荷交换[图9(B)和(C)].通过路径分析发现,ECNRR 更倾向平行氢化(后期N—N 裂解)而不是串联氢化(早期N—N 裂解).通过电催化氮还原的局部结构构筑过程进一步表明,后期N—N键的裂解起始于NH2—NH3+(H++e−),利于抑制电催化析氢的进行.通过中间体氮−氮键(即N=N和N—N)的变化实现了局部活性吸附位点对H-脱附的能量补偿[图9(D)].
Fang 等[77]通过电化学沉积的方法实现了单个锰原子在石墨炔表面的成功锚定,获得了石墨炔基锰原子催化剂(MnSA/GDY).MnSA/GDY 具有明确的最优化学结构和价态,锰原子与石墨炔之间特殊的不完全电荷转移性质显著增加了活性位点数量,为催化反应提供了丰富的活性位点,提高了材料的电荷转移能力,使其在电催化氮还原合成氨反应中表现出良好的性能(图10),氨产率达到了46.78,法拉第效率可达39.83%,大大超过了当前报道的传统原子催化剂和异质结,并且该催化剂具有较好的稳定性,循环8次后活性仍能得到保持.
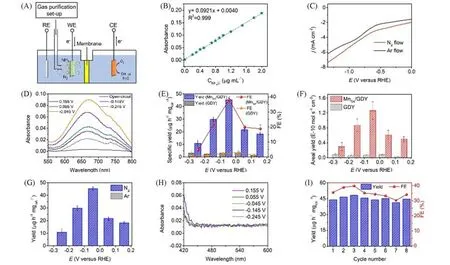
Fig.10 Schematic diagram of NRR battery configuration at room temperature(A),a calibration curve used to quantify the ammonia produced(B),polarization curves tested in nitrogen and argon atmosphere(C),UV⁃Vis spectra of electrolyte after NRR for 1 h at different potentials at room temperature(D),specific ammonia yield and Faraday efficiency of MnSA/GDY and pure GDY at different potentials in saturated N2 0.1 mol/L Na2SO4(E),area ammonia yield and Faraday efficiency of MnSA/GDY and pure GDY at different potentials in N2 saturated 0.1 mol/L Na2SO4(F),ammonia yield of MnSA/GDY ob⁃tained at different potentials in 0.1 mol/L Na2SO4 saturated with Ar and N2,respectively(the error bar represents the standard deviation)(G),in N2 saturated 0.1 mol/L Na2SO4,the UV⁃Vis spectrum of N2H4 in MnSA/GDY detected under applied potential(H),stability test results of MnSA/GDY in 0.1 mol/L Na2SO4 at-0.045 V(vs.RHE)(I)[77]
Zou 等[78]设计合成了Rh/GDY 原子催化剂,并使用自制的膜分离三电极电池,分别在常压和加压条件下检测其电催化氮气还原制氨性能(图11),发现提高体系内N2的压力能够显著提升氨产量,图11(D)显示,其在55 atm N2下,Rh/GDY原子催化剂具有很高的氨产率(74.15 μg·h-1·cm-2)和高的法拉第效率(20.36%).
图11(E)比较了不同N2压力下的最佳氨产量、法拉第效率和氨电流密度,发现随着N2压力的增加三者都显著增强,尤其在55 atm(1 atm=1.013×105Pa)下的氨产量相对于环境条件下的氨产量、法拉第效率和氨电流密度分别提高了7.3 倍、4.9 倍和9.2 倍.他们也对该催化剂的稳定性进行了测试[图11(F)],发现经过6次循环后氨产率和法拉第效率衰减很小,产氨性能保持在88.3%.这为高效电催化制氨提供了一条可行的途径.

Fig.11 Pressurized ENRR test[78]
2.2 电催化析氢反应
氢气是一种具有高能量密度的绿色能源,有望在未来替代化石能源,改善环境问题.目前主要通过电解水制氢,然而析氢反应(HER)通常需要相对较高的起始电势和较低的H2生产速率,产生巨大的电能消耗.因此,开发高效的HER电催化剂以降低反应的起始电势,加快反应速率,降低反应能耗是电解水制氢技术中最迫切的需求之一.在已报道的电催化剂中,铂基催化剂具有较好的电催化析氢性能.但是金属铂在地壳中储量稀少,使用成本高,极大限制了其大规模应用.因此,寻找具有高效、低成本的新型电催化剂具有重大的意义.原子催化剂因具有较高的原子利用率,能够有效降低使用成本.石墨炔中炔键的存在利于金属原子均匀地锚定在石墨炔的表面.因此,石墨炔基原子催化剂有望替代传统贵金属催化剂.下面将对几种已报道的具有优秀HER性能的石墨炔基原子催化剂的催化性能和稳定性等进行详细介绍.
Xue等[3]合成了石墨炔基零价Ni,Fe(Ni0/GDY 和Fe0/GDY)原子催化剂,并对它们的电催化析氢性能进行了详细的研究[图12(A)~(F)],发现相对于Pt/C催化剂,Ni0/GDY和Fe0/GDY具有更好的阴极电位响应,且Fe0/GDY的初始过电位为9 mV,与Pt/C的初始过电位(1 mV)相当.Fe0/GDY和Ni0/GDY在电流密度为10 mA/cm2时过电位分别为66 和88 mV.两种催化剂的质量活性不仅显著超过了Pt/C,而且分别具有较小的塔菲尔斜率,其中Fe/GDY 为37.8 mV/dec,Ni/GD 为45.8 mV/dec,与Pt/C(33.9 mV/dec)相当.与其它原子催化剂(如石墨烯负载的Ni原子催化剂和Co-N-石墨烯原子催化剂)相比,负载在GDY 上的零价Ni,Fe 展现了优异的电催化性能.他们还合成了负载在GDY 上的Fe 和Ni 纳米颗粒,并对其进行了电化学性能测试,发现其HER性能比相应的GDY基原子催化剂低,这也证实了GDY基原子催化剂的优势.他们也对两种原子催化剂的稳定性进行了测试,发现循环5000次后的极化曲线基本无变化,XPS和HAADF-STEM也显示金属原子仍很好地分布,没有聚集现象;同时两催化剂还展现了最大98%的法拉第效率,证实GDY对金属原子的高效锚定能力以及优异的电化学析氢性能.
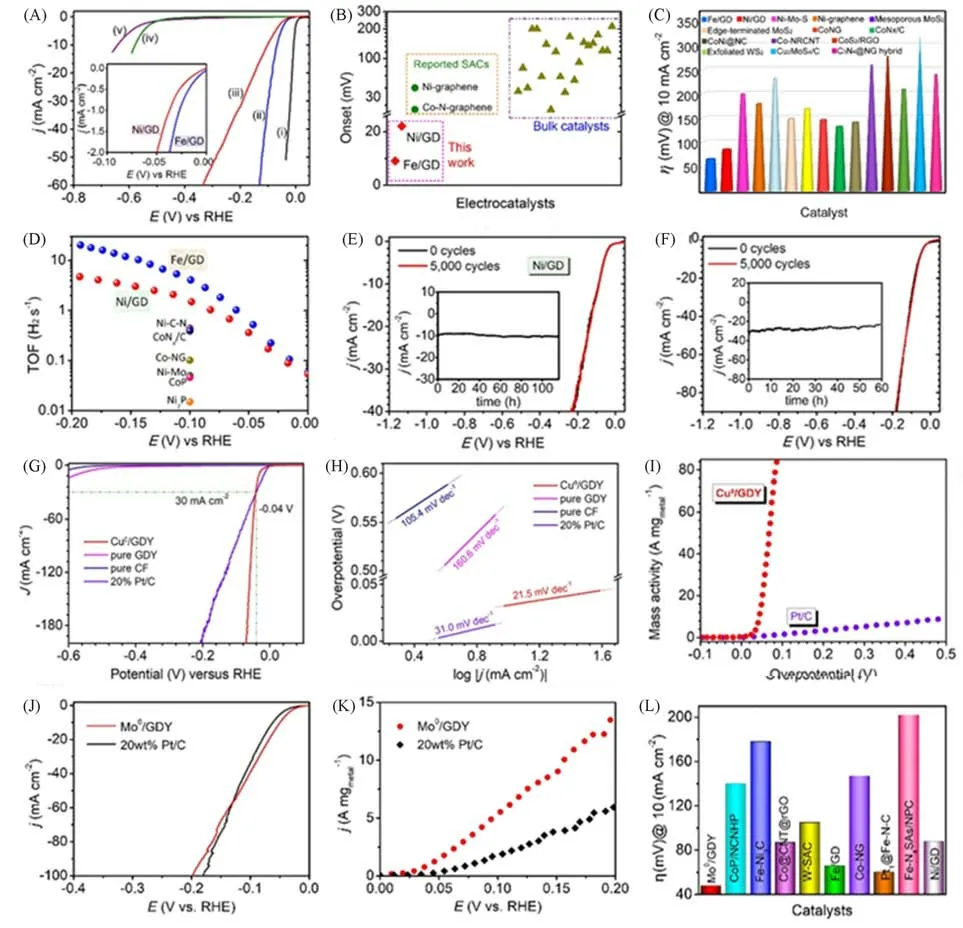
Fig.12 Polarization curves of Pt/C(I),Fe/GD(II),Ni/GD(III),GDF(IV) and CC(V) in 0.5 mol/L H2SO4 solution(inset:enlarged LSV curves of Fe/GD and Ni/GD near the starting region)(A),Ni/GD and Fe/GD(red region)and other non⁃noble metal monatomic HER catalysts(green region)and several bulk catalysts(olivine triangle) at 10 mA/cm2(B) and overpotential(C),TOF values of Fe/GD(blue dot) and Ni/GD(red dot) and several state⁃of⁃the⁃art her electrocatalysts(D),Ni/GD(E) and Fe/GD(F) [3],LSV curve(G) and corresponding Tafel slope(H),mass activity of Cu0/GDY and Pt/C(I)[4],polarization curve(J) and mass activity(K) of Mo0/GDY and 20% Pt/C,over potential of various catalysts at 10 mA/cm2(L)[5]
石墨炔负载的零价铜原子催化剂(Cu0/GDY)在0.5 mol/L H2SO4溶液中具有优异的电催化析氢性能[4].如图12所示,Cu0/GDY在10 mA/cm2的电流密度下过电位仅为32 mV,明显优于商业化的Pt/C电极及已报道的过渡金属催化剂及贵金属催化剂.为了进一步研究Cu0/GDY催化剂的催化性能,通过塔菲尔斜率研究了其催化反应动力学[图12(G)].Cu0/GDY 的塔菲尔斜率为21.5 mV/dec,明显低于Pt/C电极的塔菲尔斜率(31.0 mV/dec),表明Cu0/GDY 具有较快的反应动力学.此外,与Pt/C 电极相比,Cu0/GDY具有较高的质量活性[图12(H)].经过23 h的稳定性测试,Cu0/GDY的电催化活性基本保持不变[图12(I)].此外,如图12(J)~(L)所示,石墨炔负载的零价Mo原子催化剂(Mo0/GDY)[5]具有优异的电催化析氢的性能:Mo0/GDY 在10 mA/cm2的电流密度下过电位为48 mV,明显优于商业化的Pt/C 电极.与GDY 相比,Mo0/GDY 能够显著提高原有的电催化性能,归因于其较低的吉布斯自由能.此外,Mo0/GDY具有较小的塔菲尔斜率(33 mV/dec)和超越Pt/C(5.89 A/mgmetal)的质量活性(15.10 A/mgmetal).
Yu 等[73]采用电沉积的方法合成了Pd/GDY 原子催化剂和Pd NPs/GDY 催化剂,分别测试了它们的HER 性能,结果显示,Pd/GDY 原子催化剂在10 mA/cm2电流密度下过电位为55 mV,低于商业化20%Pt/C的62 mV,而Pd NPs/GDY 催化剂过电位为115 mV;制备的Pd/GDY原子催化剂也具有优良的质量活度和较小的Tafel 斜率;转化频率(TOF)值是单位时间内单个活性位点的转化数,被用来表示催化剂的本征活性.研究结果显示,Pd/GDY 具有最高的TOF 值,说明GDY 原子催化剂中原子具有较高的利用率.他们也考察了合成的Pd/GDY 的循环稳定性,结果显示,在循环1000 次后该原子催化剂仍具有与初始状态相当的HER 性能,可归因于GDY 与零价Pd原子之间的不完全电荷转移.Yin 等[79]通过湿化学法和退火法分别制备了两种具有不同配位环境的Pt/GDY 原子催化剂,分别命名为Pt-GDY1(C1-Pt-Cl4)和Pt-GDY2(C2-Pt-Cl2),并进行了HER性能测试,结果显示,Pt-GDY2活性最高,质量活性可达到商业化Pt/C的26.9倍;在过电位为100 mV时,Pt-GDY1,Pt-GDY2和Pt/C中,Pt-GDY2具有最高的质量活度,达到了23.64 A/mg,并且其Tafel斜率(46.6 mV/dec)比Pt/C(127.5 mV/dec)低;极化曲线显示,Pt-GDY2循环1000次后催化剂活性仍然保持,证明了其优异的稳定性.
2.3 电催化析氧反应
Yu 等[80]测试了制备的Ru/GDY 在酸性条件(0.5 mol/LH2SO4)下的析氧反应(OER)性能,发现其在10 mA/cm2下具有531 mV 的过电位,小于已报道的MnxSb1−xOz,Ni0.5Mn0.5Sb1.7Oy和NiFeP.Ru/GDY 的Tafel 斜率较小(100 mV/dec),展现了良好的酸性OER 性能.他们还将Ru/GDY 原子催化剂与Ru/GDY纳米颗粒和RuO2做对比,发现在过电位为2.0 V时,Ru/GDY原子催化剂的jmass为9.03 A/mgmetal,分别是Ru/GDY纳米颗粒和RuO2的7.05倍和451倍,远远超过了Ru氧化物,展现了原子催化剂的优势.他们还对稳定性进行了检测,发现在循环2000次后极化曲线仍能保持,且测试54 h后Ru/GDY原子催化剂的形貌、化学状态和负载量均无明显变化,这说明了锚定在GDY上的单个Ru原子非常稳定.Qi等[81]通过DFT计算了吡嗪改良的GDY(PR-GDY)锚定不同过渡金属原子形成的原子催化剂的OER性能,结果表明,Ni@PR-GDY 在OER 的4 个反应基本步骤中都有着正的自由能和较低的过电位.值得注意的是,Ni@PR-GDY具有最低的过电位(0.29 V),远远低于IrO2催化剂(0.42 V),这表明其具有优异的电催化OER性能;并且还发现,通过在TM@PR-GDY中掺杂电子或空穴,也可以通过实现电子转移进而影响催化剂的OER催化活性.在Ni@PR-GDY中,在晶胞中加减1个电子可以改变催化反应的决速步,与中性样品对比,电子掺杂的Ni@PR-GDY的决速步为OOH的吸附,这为设计GDY原子催化剂提供了理论支撑.Feng 等[82]使用DFT 模拟了单个过渡金属原子锚定在GDY 上(TM@GDY)在碱性体系中的OER 和氧还原反应(ORR)性能,构建了双火山图来描述和预测TM@GDY 对OER 和ORR 的催化活性.对于一种给定的催化剂,其OER和ORR催化性能由反应中间体的吸附吉布斯自由能决定,良好的催化剂不应对反应中间体有太弱或太强的结合.在分别对Ni@GDY和Pd@GDY,Pt@GDY进行比较后得知,Pt@GDY在碱性介质中有着最好的OER性能,其过电位为0.49 V,与RuO2(0.42 V)接近.为了研究其具有良好催化性能的内在原因,他们研究了*O 在3 种催化剂的吸附以及电荷分析,结果显示,对于3 种催化剂,Ni,Pd 和Pt 具有正电荷,促进了一些带负电的氧物种的吸附,从而起到了活性中心的作用,GDY 上捕获的过渡金属原子的价电子不仅可以与GDY 的sp2杂化C 原子的pz轨道杂化,也能与px和py杂化;此外,计算结果证明,Ni@GDY,Pd@GDY 和Pt@GDY 的扩散势垒分别为2.31,1.34 和2.52 eV,这导致了GDY锚定的过渡金属原子具有良好的稳定性.
2.4 电催化氧还原反应
Cai等[83]通过在水溶液中使用NaBH4还原吸附在GDY上的Fe3+得到Fe/GDY原子催化剂,对其在碱性条件下的ORR性能进行了测试(图13),结果表明,其具有优异的碱性ORR性能,在高电位区的Tafel斜率为63 mV/dec,与商业化Pt/C的62 mV/dec相当,并且DFT结果显示,Fe/GDY原子催化剂具有良好的选择性,对4e-ORR的电催化活性高于2e-ORR,实验验证结果与密度泛函预测值吻合.Qi等[81]通过理论计算得出,在一系列TM@PR-GDY中,Sc,Ti,V,Cr,Mn和Fe的ORR过电位大于1.23 V,不适合作为有效的ORR 电催化剂;对于Co 和Cu,其与OH 的结合较强,也阻碍了其电催化ORR 的性能.而Ni@PR-GDY 不仅拥有这种优良的OER 性能,也具有最好的ORR 性能,其电催化ORR 过电位仅为0.38 V,低于出色的Pt 催化剂(0.45 V),从而证明Ni@PR-GDY 具有良好的电催化ORR 活性.Feng等[82]通过DFT 计算得到,在碱性体系中,Ni@GDY 的过电位和Pt@GDY 相当(0.44 V),低于Pt 催化剂的过电位(0.45 V).Ni,Pd和Pt具有正电荷,促进了一些带负电的氧物种的吸附,从而起到了活性中心的作用;此外,与Ni,Pd和Pt直接相连的C原子获得了电子,增强了C-Ni,C-Pd和C-Pt的相互作用,说明锚定的金属原子与GDY基底以及催化物种之间存在电荷转移,从而增强了它们的催化活性.
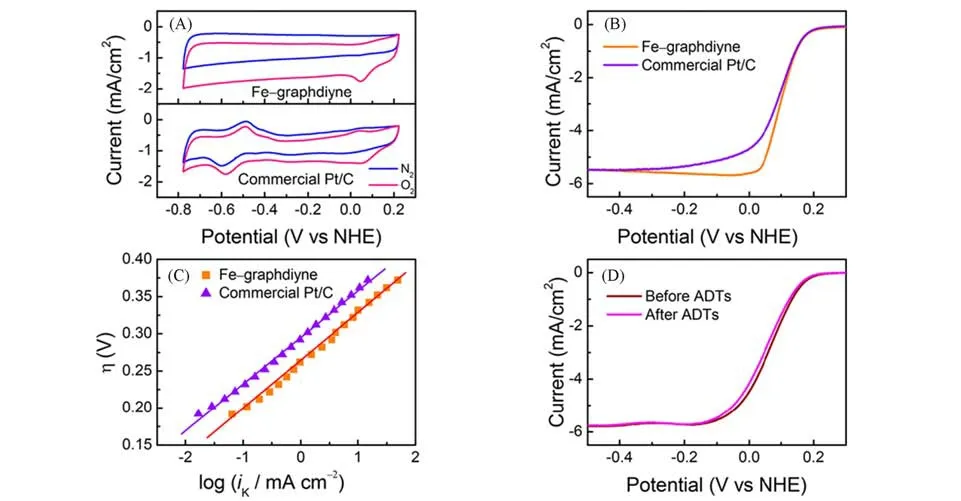
Fig.13 CV curves of Fe⁃GDY and commercial Pt/C catalysts in N2(blue line) and O2 saturated(red line)in 0.1 mol/L KOH solution at room temperature(A),RDE test of Fe⁃GDY and 20%Pt/C catalyst in 0.1 mol/L KOH solution(B),Tafel slope(C),ORR stability(D)of Fe⁃GDY[83]
2.5 电催化全水解
电化学析氢反应(ECHER)通常在酸性环境下进行,而电化学析氧反应(ECOER)在碱性条件下进行.因此,同时进行析氢和析氧的全水解(OWS)是非常困难的.目前,Ru的氧化物公认为是酸性OER的优良电催化剂,但其高昂的生产和使用成本及欠佳的稳定性限制了其进一步应用.因此,开发同时满足在酸性条件下高效HER 和高效OER 的电催化剂非常必要.Yu 等[80]使用简单的电沉积法,将Ru分散在GDY 基底上形成了一种Ru/GDY 原子催化剂,其负载量(质量分数)达1.0%.理论计算结果表明,Ru 与相邻GDY 上的C 原子产生强的p-d耦合,其中的Ru 原子化合价为+4,这有利于催化水的分解;HER性能测试结果(图14)显示,Ru/GDY原子催化剂在酸性环境下具有优异的HER性能,在10 mA/cm2下的过电位为44 mV,Tafel 斜率为30 mV/dec,在150 mV 的过电位下其质量活度(15.88 A/mg)为20%Pt/C(1.00 A/mg)的115.88 倍.此外,他们也对酸性OER 性能进行了测试,结果显示,Ru/GDY原子催化剂具有良好的酸性OER 性能,在10 mA/cm2下的过电位为531 mV,最小Tafel 斜率为100 mV/dec;同时,Ru/GDY 原子催化剂的质量活度非常高,在过电位为2.0 V 时,其质量活度(9.03 A/mg),是RuO2(0.02 A/mg)的451 倍;TOF 值(7.09 s-1)是RuO2(0.01 s-1)的709 倍;2000 次循环后CV曲线仍得到保持,超过54 h的计时电流测试也显示了Ru/GDY 原子催化剂优异的稳定性,并且形貌和负载量几乎没有变化,说明合成的Ru/GDY在酸性条件下具有非常优异的全解水性能.
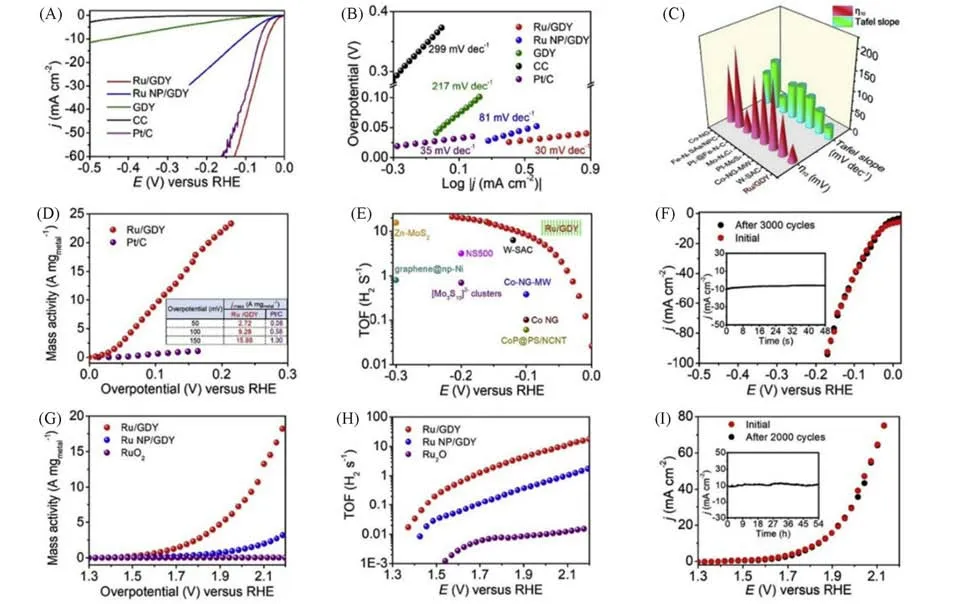
Fig.14 Polarization curve(A),Tafel slope of HER of corresponding sample(B),comparison of Tafel slopes(C),HER mass activity of Ru/GDY and Pt/C(D),comparison of TOF values between Ru/GDY and other catalysts(E) polarization curve of Ru/GDY after 3000 HER cycles(F),OER mass activity of Ru/GDY and Ru NP/GDY and RuO2(G),TOF values of OER for Ru/GDY and Ru NP/GDY and RuO2(H),polarization curve of Ru/GDY before and after 2000 OER cycles(I)[80]
He 等[84]通过理论计算得出了11 种过渡金属原子对于HER,OER,ORR 的反应活性,发现Pt/GDY和Ni/GDY 有望成为水分解的电催化剂,其中Pt/GDY 的HER 过电位为0.01 V,OER 过电位为0.29 V;Ni/GDY 的HER 过电位为0.46 V,OER 过电位为0.40 V.计算结果显示,原始GDYC 位的ΔGH*为0.73 eV,表明C原子与活性物种结合能力很弱.而Pt/GDY的ΔGH*非常接近最佳值(0 eV),这是因为GDY吸附Pt 后电荷重新分布,增强了C 与活性物种的相互作用.Ni/GDY 和Pt/GDY 在OER 中的ΔGOH*分别为0.29 V和0.46 V,可能成为高性能的全解水电催化剂.
2.6 电催化CO2固定
当前,CO2的大量排放造成了一系列环境问题,阻碍了人类社会的发展,各国都在致力于CO2排放的限制与CO2封存.我国更是提出了碳中和战略,可见CO2的固定是非常重要的.而将CO2转化为人类需要的化工产品是一条有潜力的途径.电催化CO2还原是利用可再生电力将CO2转化为人们需要的物质,但CO2的化学性质惰性,导致转化反应较难进行.一直以来,人们致力于开发高效催化CO2转化的各种催化剂,但尚未取得很满意的结果.如,作为均相催化剂的离子液体基催化剂和金属配合物催化剂虽然具有良好的选择性,但面临着难以分离和难以回收利用的缺点;作为异相催化剂的MOFs等虽然易于分离,但合成条件苛刻和催化剂性能欠佳等缺点限制了其进一步应用.因此,开发一种低成本高效的CO2转化催化剂是非常必要的,而GDY基原子催化剂展现了其独特的性质以及其高效的CO2转化性能.Zheng等[85]报道了一种基于电沉积的GDY基零价Co原子催化剂,该催化剂在CO2与环氧苯乙烯的环加成反应中表现了相当高的转化率和选择性(图15),在80 ℃时对环氧苯乙烯的转化率接近100%,并且选择性高达99%以上;并且,零价Co/GDY表现了高达3024.8 h-1的TOF值,显著高于之前报道的催化剂;并且该催化剂回收简单,只需乙酸乙酯洗涤,性能可保持15次.DFT 结果显示,正是环氧苯乙烯的氧原子与Co/GDY 上的Co原子之间发生了强电荷转移,从而导致了Co/GDY 易于吸附环氧苯乙烯,有利于随后的开环反应.产物的脱附是放热过程,有利于完成整个催化过程.Liu等[86]通过DFT计算了基于GDY基过渡金属原子催化剂的CO2还原催化性能,得到不同GDY基原子催化剂对CO2还原的催化性能和产物的选择性不同,其中Co/GDY倾向于生成CH3OH,Fe/GDY倾向于生成HCOOH.这是由于过渡金属原子的d 带中心与CO2还原的中间产物的吸附越强,催化性能越好.Co/GDY 有着−0.64 eV的能带中心,并且对氧中间体的结合较强,故Co/GDY有望成为电催化CO2还原的催化剂.He等[87]通过DFT计算得出,在GDY上含有不同数目Fe的Fe/GDY原子催化剂对CO2还原的产物选择性不同,其中Fe 二聚体对CO2转化为CH4的催化活性最高,决速步为0.29 eV;Fe 三聚体对CO2转化为HCOOH的活性最高,决速步为0.35 eV.并且计算结果还表明,在GDY上沉积的单个Fe原子向另一个稳定位置移动的扩散能垒高达3.41 eV,这表明GDY 上Fe 的聚集是很难的,从而保证了Fe/GDY 原子催化剂的稳定性.

Fig.15 Time course of catalytic conversion at different temperatures(A),TOF value and yield of Co0/GDY at different temperatures(B),compared with the reported catalysts(C),comparison of SO conversion between Co0/GDY and reported catalysts(D),stability test of catalyst(E),under the same conditions,carbonates with different substituents generated from the corre⁃sponding epoxy compounds under the catalysis of Co0/GDY(F),free energy distribution of key intermediates(G),Co0/GDY and SO adsorbed Co0/GDY Co⁃3d PDOS(H),differential charge density diagram of SO adsorbed Co0/GDY(I)[85]
2.7 其它应用
除了以上介绍的应用,GDY原子催化剂在其它领域也大放异彩.Yin等[88]制备了Pd/GDY原子催化剂并用于炔烃的半加氢制备烯烃反应,结果表明,Cu/GDY 原子催化剂在−1.0 V时对甲烷的选择性较C2产物高,且法拉第效率达到57.3%;Ali等[89]通过DFT模拟Au/GDY原子催化剂用于乙炔的氢氯化反应,结果证实,在GDY上负载的单个Au原子较在石墨烯和单壁碳纳米管上具有更高的性能;Xu等[90]通过DFT研究了Rh/GDY原子催化剂用于O2氧化CO的反应,发现Rh可以稳定地存在于GDY上,并且对O2和CO 分子具有强的吸附能力,是一种有潜力的CO 氧化的催化剂;Sun 等[91]用DFT 研究了4 种M/GDY 原子催化剂(M=Pt,Rh,Cu,Ni)的CO 氧化活性,发现无论是根据计算的反应能垒还是根据各反应机理的初态能量,Cu/GDY原子催化剂都被预测是一种高效催化剂.Xu等[92]计算了Ir/GDY原子催化剂用于氧化CO的不同机理的反应能垒,发现CO的氧化更倾向于Eley-Rideal(NER)机理,而反应的决速步是吸附的O2分子被2分子CO活化形成两分子CO2,反应能垒是0.37 eV;Zhang等[93]将单个Pd原子锚定在GDY/石墨烯异质结上,并用于催化4-硝基苯酚转化为4-氨基苯酚,该催化剂表现出了较高的催化活性和选择性,反应速率常数达到了Pt/C 的44倍.这些充分说明了GDY 原子催化剂在其它应用领域也具有广阔的发展前景.
3 总结与展望
自2010年石墨炔被成功合成以来,已在众多领域发挥出重要作用.得益于其独特的结构和性质,石墨炔在零价金属原子催化剂的合成与应用方面具有天然优势.石墨炔零价原子催化剂的出现解决了传统单原子催化剂化学/电子结构不明确、金属原子易团聚等问题,为解决诸多领域的关键问题提出了崭新的思路和理念,催生了一系列具有独特功能和变革性质的催化剂.石墨炔原子催化剂具有合成条件温和、可大规模快速合成等优势,使原子催化剂的商业化应用成为可能.石墨炔原子催化剂的出现为设计与制备新型高效催化剂提供了新思路.
虽然石墨炔原子催化剂的研究成果显著,但目前仍处于起始阶段,仍需深入探索其重要的功能应用:(1)拓展石墨炔基原子催化剂合成的新方法,实现宏量制备;(2)探究金属原子与载体之间的电子转移机理和相互作用关系,实现对金属原子锚定过程和锚定位点的精确可控调控;(3)基于石墨炔原子催化剂带来的新性质、新现象和新效应,实现其在不同研究领域的新功能和新应用.这些方面的实现将为开发具有我国自主知识产权的石墨炔基新型催化材料、推动我国石墨炔科学发展做出重要贡献.
猜你喜欢
物理化学学报(2024年9期)2024-09-27 00:00:00
江苏安全生产(2023年1期)2023-02-08 05:57:50
江苏安全生产(2022年6期)2022-07-29 01:22:32
环境工程技术学报(2022年3期)2022-06-05 07:20:34
江苏安全生产(2021年5期)2021-07-16 06:47:16
原子与分子物理学报(2021年1期)2021-03-29 07:29:14
四川环境(2018年3期)2018-06-28 03:43:26
中国有色金属学报(2018年2期)2018-03-26 07:58:37
中南大学学报(自然科学版)(2016年2期)2017-01-19 07:37:25
表面工程与再制造(2016年5期)2016-12-15 11:42:26
