
Synthesis of rac-α-aryl propionaldehydes via branched-selective hydroformylation of terminal arylalkenes using water-soluble Rh-PNP catalyst
2022-06-18 03:00PengGoMiolinKeTongRuGunfengLingFenErChen
Chinese Chemical Letters 2022年2期
Peng Go, Miolin Ke, Tong Ru, Gunfeng Ling,*, Fen-Er Chen,,*
a Department of Chemistry, Sichuan University, Chengdu 610064, China
b Department of Chemistry, Fudan University, Shanghai 200433, China
c Shanghai Engineering Center of Industrial Asymmetric Catalysis for Chiral Drugs, Fudan University, Shanghai 200433, China
ABSTRACT This work detailed the preparation of a class of water-soluble PNP ligands that differed by the nature of the substitute on phenyl ring of ligands.These ligands were incorporated into water-soluble rhodium-PNP complex catalysts that were used to regioselective hydroformylation of a series of terminal arylalkenes,providing efficient access to rac-α-aryl propionaldehydes in good to excellent yield (up to 97%) and branched-regioselectivity (up to 40:1 b/l ratio).Furthermore, gram-scale and diverse synthetic transformation demonstrated synthetic application of this methodology for non-steroidal antiinflammatory drugs.
Keywords:Aryl propionaldehydes Hydroformylation Water-soluble PNP ligands Regioselectivity Rhodium
rac-α-Arylaldehydes have been extensively used as important synthetic precursors for the profen family (α-arylpropionic acid derivatives), a class of non-steroidal anti-inflammatory drugs(NSAIDs) in pharmaceutical chemistry [1-5].At present, more than 20 profen NSAIDs has been used in clinical for treating the heat of inflammation, pain, swelling and fever [6,7].Some representative examples are illustrated in Fig.1, such as ibuprofen (1) [8], ketoprofen (2) [9], flurbiprofen (3) [10], naproxen (4) [11] and loxoprofen (5) [12].Undoubtedly, the development of efficient approaches towardsrac-α-arylaldehydes is always a popular topic in both academic and industrial laboratories [13-17].Traditional methods for synthesis ofrac-α-arylaldehydes include the rearrangementviaDarzen’s glycidic esters [18,19] and Corey-Chaykovsk’s epoxides [20] from aryl methylketones (Schemes 1a and b).However,these processes suffer from several drawbacks such as more complicated synthetic operations, harsh reaction conditions, and functional group incompatibility.Attractive alternatives have been developed using 1,1-arylmethyl alkenes as feedstocks (Scheme 1c)[21-23], including Che’s Ru(IV) porphyrin-catalyzed aerobic oxidationviaan epoxidation/isomerization tandem pathway [21], and Lahiri’s catalytic PhIO oxidation by applying iron(II) dipic catalyst,in situformed from Fe(BF4)2·6H2O and pyridine-2,6-dicarboxylic acid [22], and Lei’s anti-markovnikov oxygenation with H2O as oxidant by a photoredox-metal dual catalysis [23].However, protocols currently lack broad substrate scope due to the difficulties for access to 1,1-arylmethyl alkenes as special substrates.
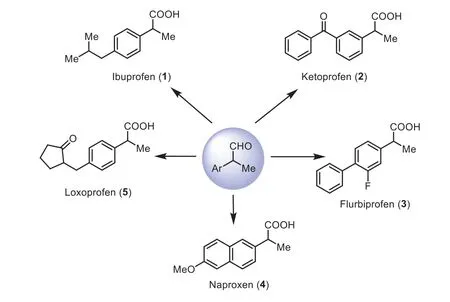
Fig.1.Representative examples for synthesis of rac-profen family (1-5) from the corresponding rac-α-aryl propionaldehydes.
The transition metal-catalyzed hydroformylation of terminal arylalkenes to arylaldehydes could provide an alternative, however typically only a small percent of theβ-arylaldehyde products was formed alongside theα-arylaldehyde products.There are remarkable exception of Rh phosphine catalysts [24-35] including Wink’s a cationic bis(dioxaphospholane)rhodium complex [24], Marchetti’s Rh/pyridylphosphines pydiphos P-oxide complex [25], Amer’s Rh/amino phosphine complex [26], Alper’s Rh/diphenylphosphinoyl)phenylmethanol catalysis [27], Börner’s Rh/diphosphate catalysis[28], Lerous’s Rh/dibenzophosphole catalysisetc.[29], which form theβ-aldehyde products with a good level of selectivity, but only for very limited substrates (Scheme 1d).Recent branched-selective hydroformylation of terminal arylalkenes has been reported using PPh3-modified Fe as catalyst [30], but the limited success has been achieved toward this hydroformylation (Scheme 1d).Hydroformylations of terminal olefins were independently reported by Nozaki’s group and Clarke’s group, but the two methods were limited to the preparation ofα-alkylaldehyde products [31,32].Apart from this class of homogeneous catalysis, significant breakthrough have been achieved toward synthesis ofβ-arylaldehydesviabiphasic Rhcatalyzed hydroformylation of terminal olefin using water-soluble poly(4-pentenoic acid (PPA)/bis(2-diphenylphosphino)ethyl) (DPPEA) or tri-meta-sulfonatophenylphosphine (TPPTS), or phenyl-β-D-glucopyranoside catalyst as ligands (Scheme 1e) [36-38].Despite the obtained achievements in this field, the developing of a practical and efficient approach to access therac-α-aryl propionaldehydes is still highly desirable from the perspective of green, step and atom economy [39].Herein, we report the preparation of a series of novel water-soluble PNP ligands and their applications to the Rh-catalyzed branched-selective hydroformylation of terminal arylalkenes for the synthesis ofrac-α-aryl propionaldehydes in aqueous biphasic catalytic system.
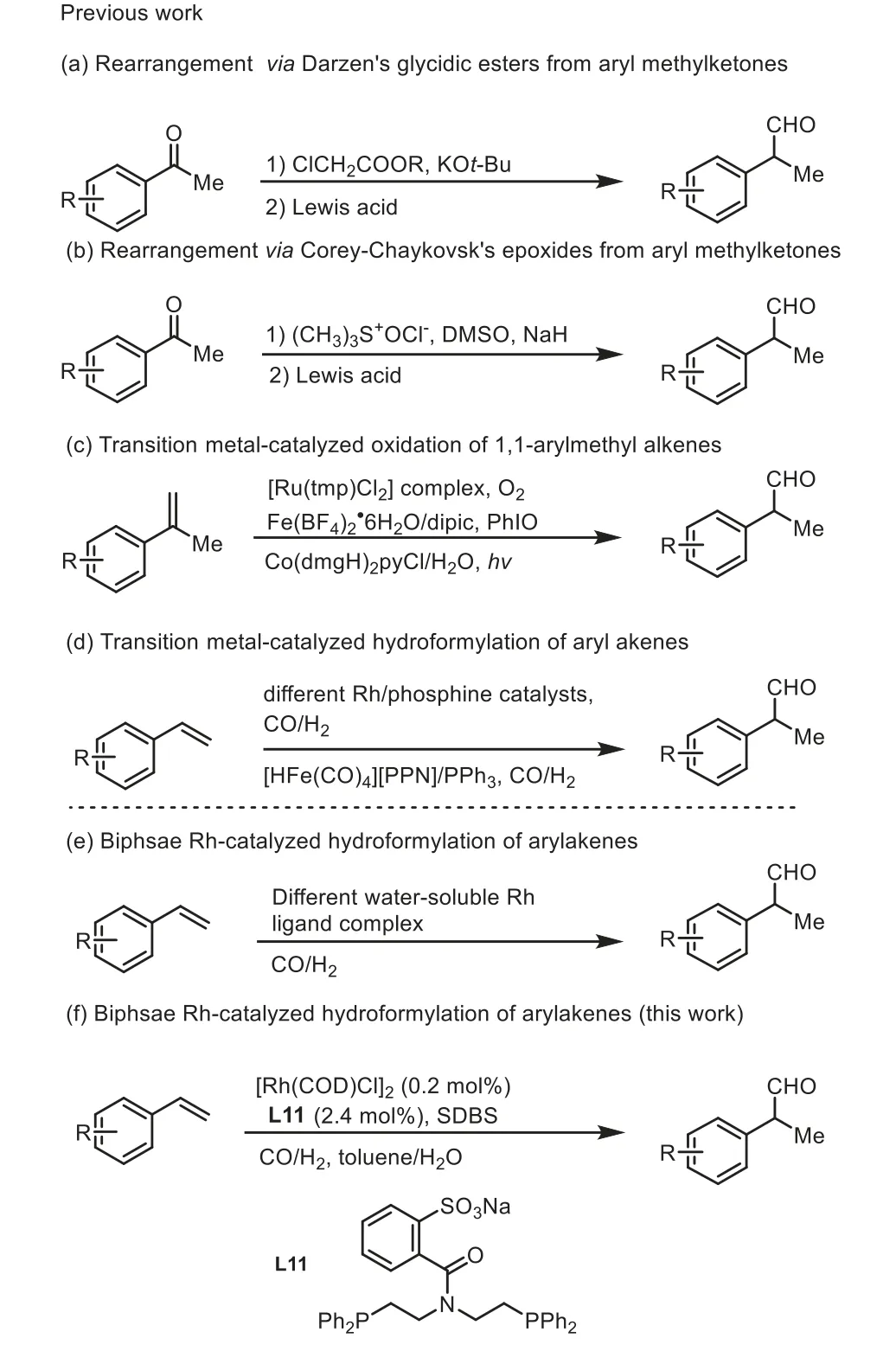
Scheme 1.Strategies to access rac-α-arylaldehydes.
The known water-soluble PNP ligands L5 and L11 and new ligands L1-L4 and L6-L10 were prepared in high to excellent yields from bis(2-diphenylphosphinoethyl)amine hydrochloride (6) and appropriate carbonyl halides, and diphenic anhydride (9) and 2-sulfobenzoicanhydride (10) following a modified procedure developed by Whiteside and co-workers (Fig.2) [40].
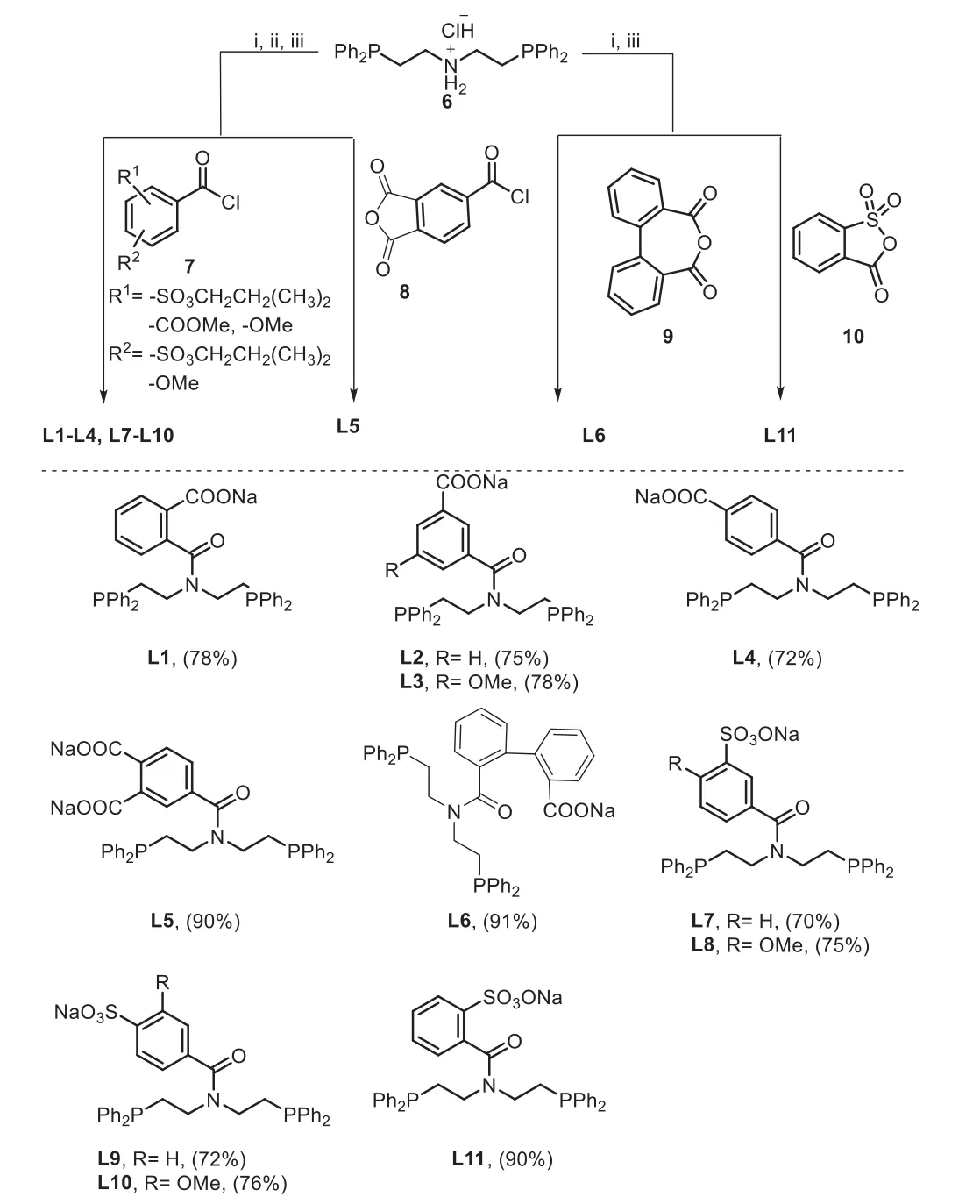
Fig.2.Synthesis of water-soluble PNP ligands (L1-L11).(i) TEA, THF, r.t., 20-24 h;(ii) NaI, acetone, reflux, 20-30 h, or LiOH, THF/H2O, 4-20 h; (iii) NaOH, THF/H2O,0.5-1 h.
With this library of water-soluble PNP ligands in hand, we began our studies with an examination of their catalytic performance in Rh-catalyzed branched-selective hydroformylation of styrene 11a as the benchmark substrate.As described in Table 1, all styrene hydrorformylation reactions were completed in 24 h toluene/H2O(1:1) solvent at 3.0 MPa of syngas and 60 °C in the presence of 0.05 mol% [Rh(COD)Cl]2and 0.6 mol% ligand (Rh/L = 1:6).In all the excellent chemoselectivities were observed, and no hydrogenation product was detectedvia1H NMR analysis.The carbonated PNP ligands L1-L6 were first examined.The ligands L1 and L2 bearing COONa at benzene’sortho- ormeta-position, gave the branched aldehyde 12a in moderate yields with good regioselectivities (b/l= 8.5:1 and 8.4:1, entries 1 and 2), respectively.When the ligand L3 substituted withmeta-OMe andmeta-COONa groups on phenyl ring was employed, poor yield of 12a was obtained, however, theb/lratio in this instance was still exceptional (8.4:1, entry 3).Reaction conducted with the ligand L4 possessing COONa group atpara-position of phenyl ring, delivered branched aldehyde 12a in only 42% yield and with lower regioselectivity (b/l= 6.5:1, entry 4).The dicarboxylated PNP ligand L5 gave better yield (75%, entry 5)compared with results of monocarboxylated PNP ligands (L1-L4),but with poor regioselectivity (b/l= 3.0:1).Moreover, the monocarboxylated PNP ligand L6 with a biphenyl ring resulted in much higher yield (84%), albeit with a ratio of 12a/13a of 5.4:1 (entry 6).The sulfonated PNP ligand L7 withmeta-SO3Na gave poor yield and regioselectivity (26%,b/l= 3.8:1, entry 7) under identical conditions.The ligand L8 bearingmeta-SO3Na andpara-MeO groups of phenyl ring was shown to give superior yield of 12a, but much worse regioselectivity (12a/13a = 3.2:1) was obtained (entry 8).A change of the position of SO3Na group on ligand L7 frommeta- topara-position resulted in similar yield (entry 9), but high regioselectivity compared with the results of the corresponding sulfonated PNP ligand L7-L10.The use of ligand L11 withortho-SO3Na group on phenyl ring afforded branched aldehyde 12a in 92% yield with high regioselectivity (b/l= 11.3:1, entry 11).The well-known TPPTS only gave 74% yield of 12a, with poor regioselectivity (b/l= 2.8:1,entry 12) under identical conditions.Other common Rh catalysts for hydroformylation, such as Rh(acac)(CO)2) and RhCl3, low regioselectivities and reactivities were respectively obtained (entries 13 and 14).Addition of SDBS (0.86 mol%) as the surfactant could short reaction time to 12 h, and provide branched 12a in 91% yield and a 10.1:1 ratio ofb/l(entry 15).Branched product was obtained in 72% yield with 10.0:1 ratio ofb/lin absence of SDBS (Table 1,entry 16).The control experiment of absence of ligand was also carried out to give the product in 74% yield with 2.8:1 ratiob/l(entry 17).These results revealed the crucial role of these two types of ligand structures in controlling of yield and regioselectivity for this Rh-catalyzed hydroformylation reaction.
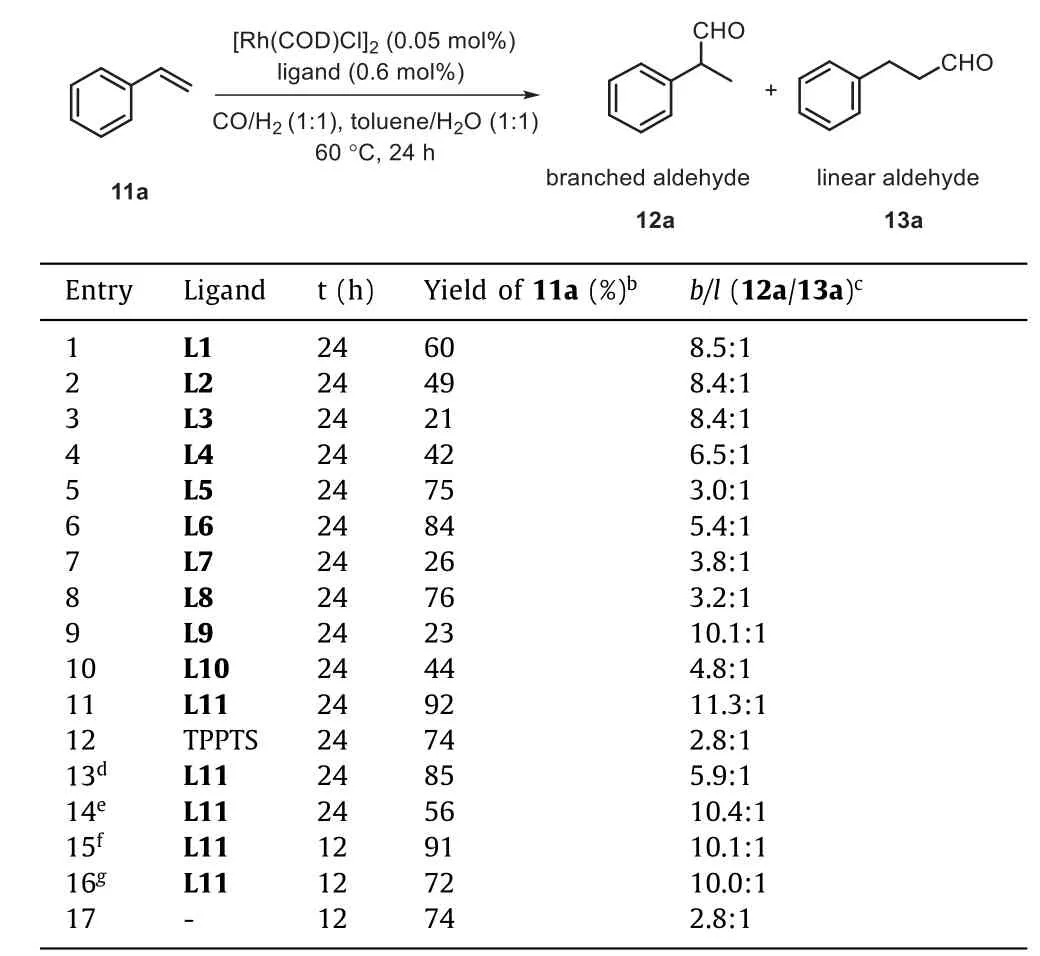
Table 1 Ligands screening for the Rh-catalyzed branched-selective hydroformylation of styrene.a
With the best ligand L11, a series of Rh-catalyzed styrene hydroformylation parameters were examined, including temperature,Rh/ ligand L11 molar ratio, organic/aqueous biphasic system, and CO/H2pressure (Table 2).First, the temperature impacted on theactivity and regioselectivity of the Rh-catalyzed styrene hydroformylation was investigated.Notably, the activity and regioselectivity of the reaction had obvious temperature effect.Lowering the temperature from 60 °C to 25 °C under standard condition(toluene/H2O (1:1), 3.0 MPa CO/H2(1:1), 0.86 mol% SDBS) dramatically reduced the yield to 8% within 12 h (entry 1vs.2).Performing the reaction at 50 °C revealed an improved yield of 70% with good regioselectivity (b/l= 11.1:1, entry 3) with full conversion in 12 h.At temperature over 60 °C, the regioselectivity and yield for the branched 12a was also significantly reduced (68%,b/l= 2.1:1 at 70 °C; 60%,b/l= 1.6:1 at 80 °C; entries 4 and 5).Increasing or decreasing the molar ratio of Rh/ligand L11 distinctly impaired the yield and regioselectivity for this transformation.A decrease of the Rh ligand L11 molar ratio from 1:6 to 1:3 led to low regioselectivity (b/l= 3.7:1, entry 1vs.6).Moreover, further increasing the molar ratio of Rh/ligand L11 to 1:8 produced a significant rise in regioselectivity (b/l= 9.7:1), but with poor yield (47%, entry 7).Varying pressure of syngas (CO/H2= 1:1) did not retard the rate but did alter the selectivity towards branched-selective product.Reducing the syngas pressure to 2.0 MPa resulted in a decrease in branched aldehyde 12a fromb/l= 10.9:1 to 7.9:1 (entry 1vs.8).A slightly increase in yield and regioselectivity was observed when 4.0 MPa syngas pressure was used for this transformation (entry 9).In addition, the use of other organic/aqueous biphasic systems,suh as DCM/H2O, MTBE/H2O, hexane/H2O, cyclohexane/H2O, was found to be less effective than toluene/H2O (entries 10-14).After reaction conditions were screened, the Rh-catalyzed hydroformylation reaction was performed using L11 as a ligand under 4.0 MPa of syngas (CO/H2, 1:1) at 60 °C in toluene/H2O (1:1) biphasic system in the presence of 0.86 mol% SDBS.Optimized conversion, yield for branched aldehyde 12a, and regioselectivity were achieved under this reaction conditions.The water-soluble Rh/L11 catalyst can be readily recycled by simple phase separation and still gave 88% yield of 12a in the second run (entry 15).
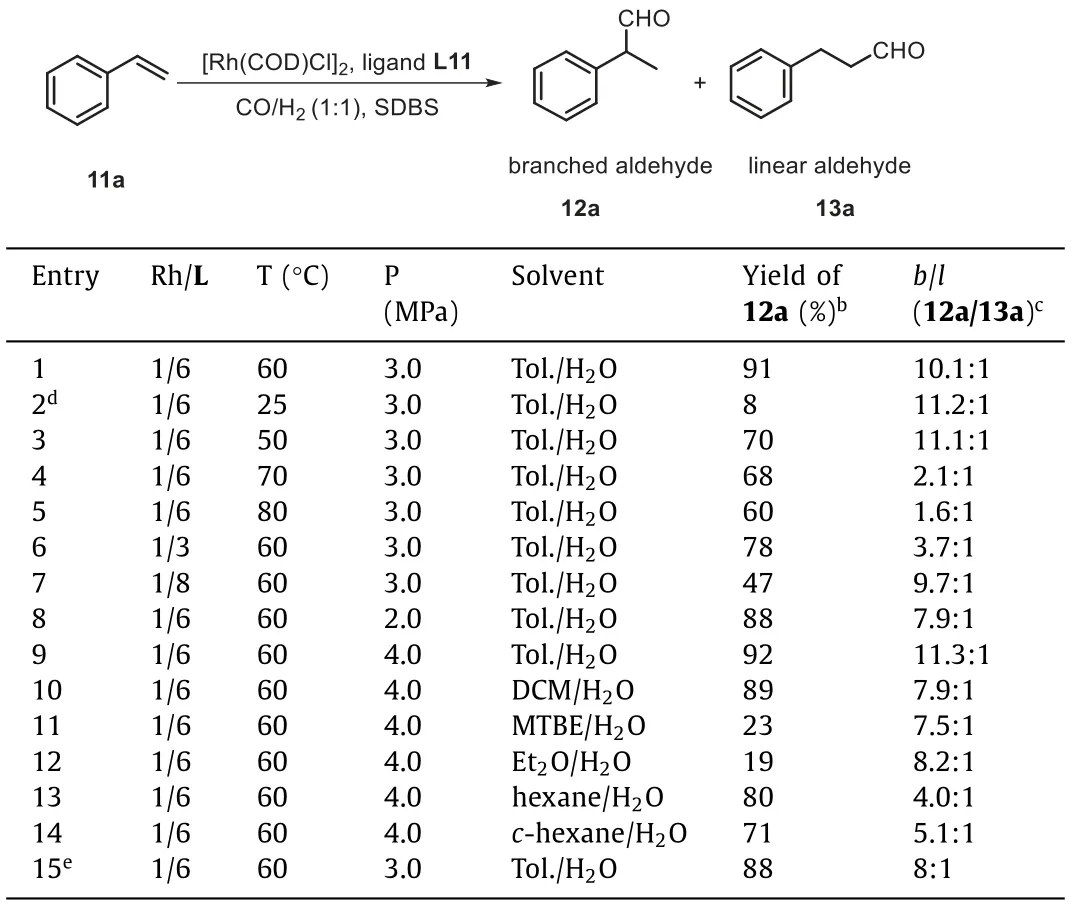
Table 2 Optimization of the Rh-catalyzed hydroformylation of styrene using L11 as the ligand.a
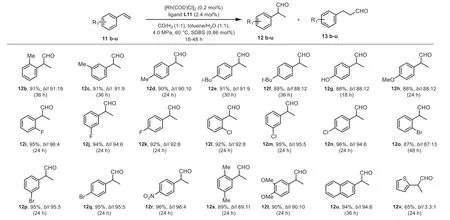
Scheme 2.Scope for the hydroformylation catalyzed by Rh/L11.Reaction conditions: Substrate (0.5 mmol), [Rh(COD)Cl]2 (0.2 mol%), L11 (2.4 mol%), toluene (1 mL), H2O (1 mL), 60 °C, 18-48 h, syngas (CO/H2 = 1), SDBS (0.86 mol%), all yields were isolated yield.
With the optimized reaction conditions, the suitability of this protocol was investigated with a range of terminal arylalkenes.As shown in Scheme 2, the mono-substituted phenyl terminal alkenes bearing electron-donating (methyl (11b-11d),iso-butyl (11e),tertbutyl (11f), hydroxyl (11g), methoxy (11h)), electron-withdrawing(fluro (11i-11k), chloro (11l-11n), bromo (11o-11q), and nitro (11r))groups at any of the position of phenyl ring were hydroformylated efficiently to give the branched aldehydes (12b-12r) in 83%-94% yield withb/l= 6.9:1-26:4) with full conversion within 24-48 h.Generally, the mono-substituted phenyl terminal alkenes bearing electron-withdrawing groups exhibited higher reactivity and regioselectivity than their electron-donating groups (11i-11rvs.11b-11h).Moreover, several sensitive functional groups including chloro (11l-11n), bromo (11o-11q), and nitro (11r) were well tolerated, no hydrogenolysis or hydrogenation products was detected by GC analysis.Similarly, disubstituted phenyl termial alkenes with electron-donating (dimethyl 11s, dimethoxy 11t) groups were also smoothly hydroformylated into the branched products 12s-12t in 86% and 90% yield with good regioselectivity (b/l= 8.1:1 and 9.1:1)resepectively.In addition to phenyl terminal alkenes, 2-naphthyl terminal alkene (11u) was also a suitable substrate for this hydroformylation reaction, delivering the branched product 12u in 89%yield withb/l= 15.1:1.The heterocylcic olefin bearing thienyl ring also smoothly transformed to the corressponding product 12v in 65% yield along with low selectivity (b/l= 3.3:1).
To further evaluate the efficiency and synthetic utility of this branched selective hydroformylation, we carried out the scale-up experiment using 6 mmol (1.10 g) of 6-methoxy-2-vinylnaphthalene 11w [41], which could be easily prepared in two steps with an overall yield of 50% starting from commercially available 6-methoxy-2-acetonaphthalene (14).The branchedselectivehydroformylation of 11w could be achieved using 0.2 mol%of the Rh/L11 catalyst within 24 h under standard reaction conditions (4.0 MP of CO/H2(1:1)) at 60 °C in tulene/H2O (1:1) to give the aldehyde 12w in 97% yield withb/l= 40:1.Oxidation of crude product 12w under Pinnick reaction conditions (NaClO2, KHPO2,2-methyl-2-butene,t-BuOH, H2O, 0 °C to r.t., 1 h) providedracnaproxen (4) in 90% isolated yield (Scheme 3) [42].
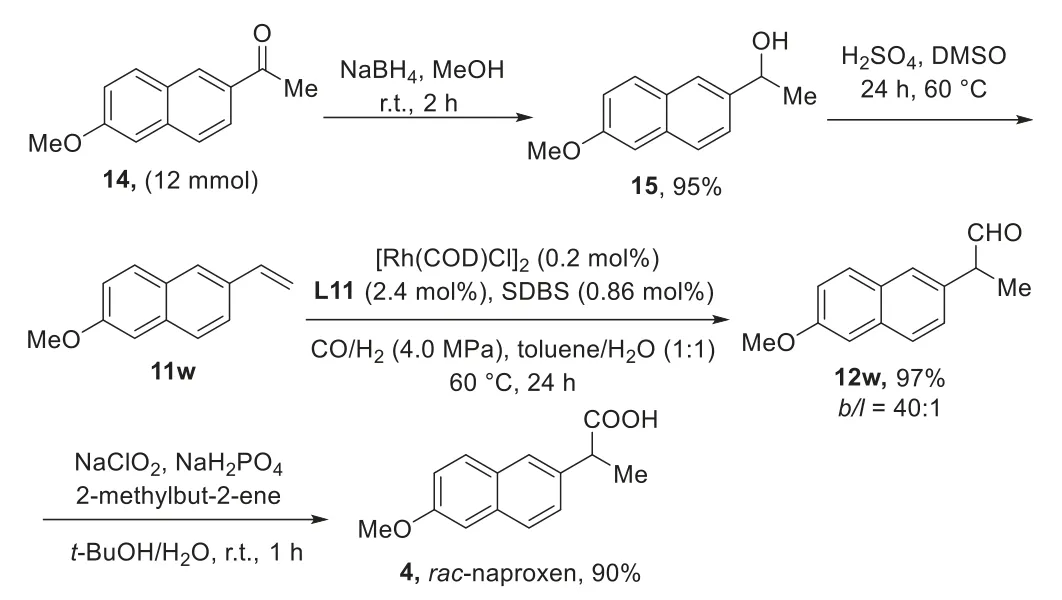
Scheme 3.Gram-scale synthesis of rac-naproxen (4).
In conclusion, we prepared a series of new water-soluble PNP rhodium catalysts and screened their catalytic performance in biphasic aqueous hydroformylation of vinyl arenes.A benzenesulfonates-containing PNP ligand L11 gave the most effi-cient Rh complex catalyst, affording high regioselectivities towardsα-aryl propionaldehydes from the branched-selective hydroformylation of various terminal arylalkenes.The exploration of substrate scope showed that the vinyl arenes with electron-withdrawing groups were usually more branched selective than those bearing electron-donating groups.Finally we demonstrated an aqueous route to the synthesis of anti-inflammatory drugsrac-naproxen in an efficient and green manner.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We are grateful for financial support from the National Natural Science Foundation of China (No.21902032).G.Liang acknowledges the funding support from Fudan University.
 Chinese Chemical Letters2022年2期
Chinese Chemical Letters2022年2期
- Chinese Chemical Letters的其它文章
- Comment on “Acid-induced tunable white light emission based on triphenylamine derivatives”
- Strategies for efficient photothermal therapy at mild temperatures:Progresses and challenges
- Liposome-based delivery of biological drugs
- Macrophage-targeted nanomedicine for chronic diseases immunotherapy
- Advances, opportunities, and challenge for full-color emissive carbon dots
- Fluorine-containing agrochemicals in the last decade and approaches for fluorine incorporation